



无菌药品作为高风险产品,其在生产管理和质量管理上具有其高标准和特殊性,欧盟《人用和兽用药品生产质量管理规范指南》附录1“无菌药品生产”(EU GMP附录1) 的发布对无菌药品的生产提出了新的要求和广泛的影响,上海医疗测试仪器展业内人士认为了解和掌握我国《药品生产质量管理规范(2010年修订)》附录1“无菌药品”(中国GMP附录1)与EU GMP附录1的差异不仅可以为无菌药品质量管理提供新的思路,也可以为中国本土产品的出海明确了相关要求差距。
01 整体章节差异:
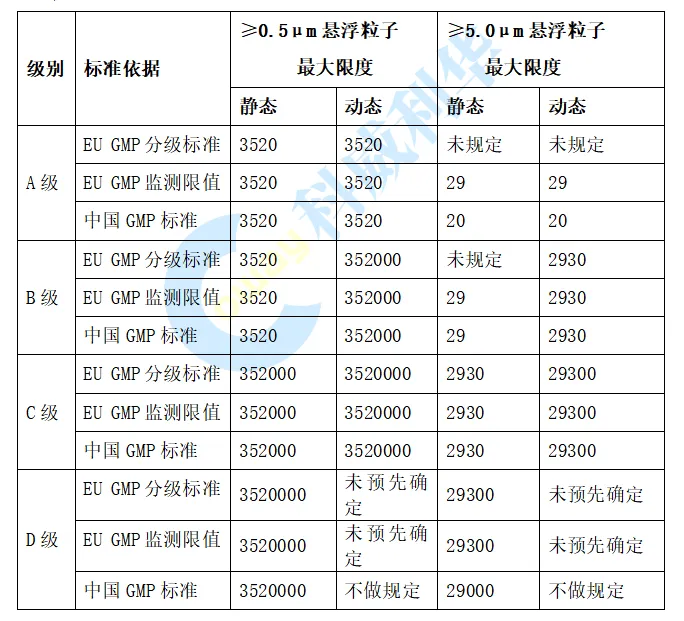
图片来源:
在微生物限度方面,在中国GMP 附录1仅规定在动态环境监测中进行微生物监测,EU GMP 附录1要求在洁净室确认以及环境监测中均需要进行微生物监测。中国GMP规定A级的微生物监测的标准为<1 cfu,此标准在执行过程中存在如果环境监测有微生物生产,但监测结果的平均值<1 cfu,检测结果是否符合要求的争议; 欧盟GMP将A级微生物标准修订为“无生产”,消除了标准的异议。欧盟与中国GMP各级别环境微生物限度标准对比见下表:
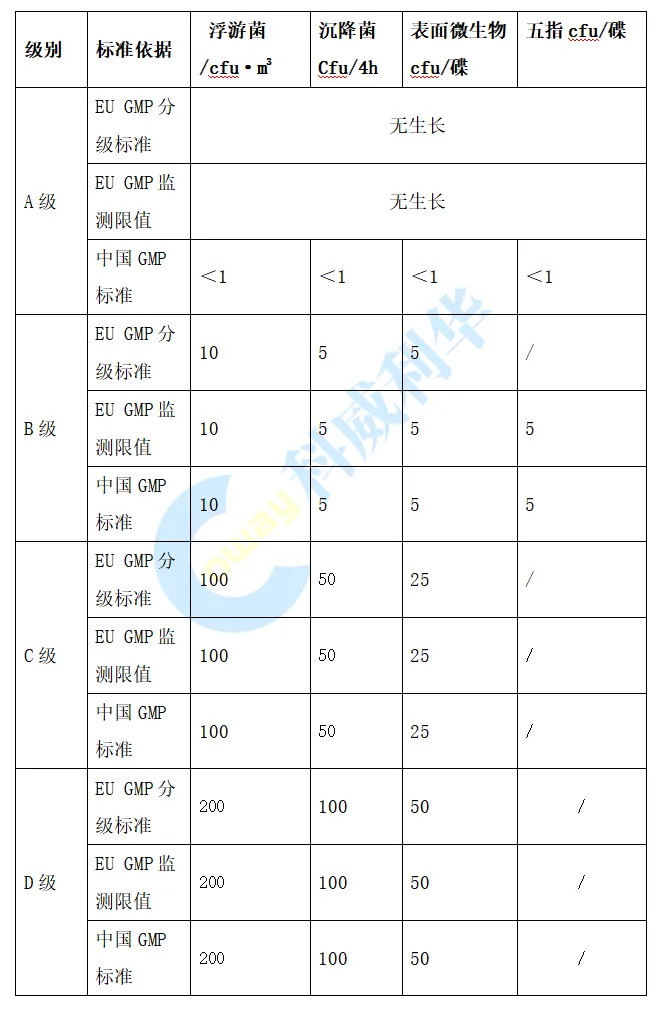
图片来源:
文章来源:
若涉及侵权,请立刻联系删除
关键字:


