



医疗质量检测技术展|中国GMP医疗器械生产质量管理规范包含哪些?
生产企业应当按照医疗器械生产质量管理规范的要求,结合产品特点,建立健全与所生产医疗器械相适应的质量管理体系,并保证其有效运行。在医疗质量检测技术展上,这些生产企业将有机会展示他们如何遵循这些规范,并通过先进的质量管理系统确保其产品的高标准和可靠性。
上海医疗测试仪器展|欧盟与我国GMP无菌药品附录差异分析

无菌药品作为高风险产品,其在生产管理和质量管理上具有其高标准和特殊性。
医疗质量检测技术及测试仪器展|医疗器械GMP要升级

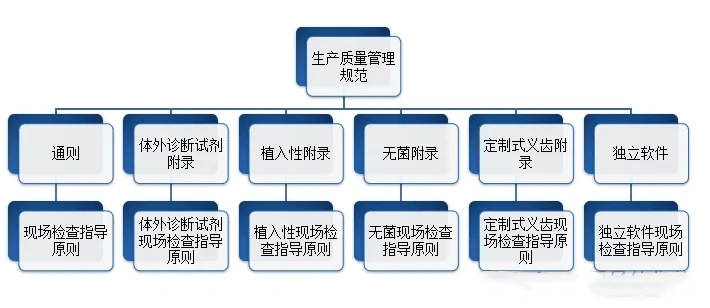
2014年12月,原国家食品药品监督管理总局(CFDA)发布了医疗器械生产质量管理规范(以下简称医疗器械GMP)(2014年第64号),并随后陆续推出了无菌医疗器械、植入性医疗器械、体外诊断试剂、定制式义齿和独立软件(注:NMPA发布)等5个附录。
医疗质量检测技术及测试仪器展|医疗器械GMP:检验设备是否都需要校验

企业生产医疗器械时,为确认产品性能指标以及生产和检验环境的洁净度,需要对产品性能指标、洁净区环境、工艺用水、产品无菌性能、环氧乙烷残留量(EO灭菌)等进行检验。质量检验的结果是否准确可靠,取决于检验设备测量的结果的准确性,企业在使用检验设备时,需要提供客观证据,证明设备符合要求。
医疗质量检测技术及测试仪器展|医疗器械GMP对“空调净化系统”的要求

医疗器械的生产过程和加工工序必须在洁净室(区)内进行,并达到规定的洁净度级别要求,不同医疗器械适用的洁净度级别要求不一致。对洁净室(区)的工作环境必须严格控制,才能有效地防止工作环境对医疗器械的污染,保证产品质量和使用者安全。而洁净度级别的控制主要通过空调净化系统的科学合理选择、设计、维护实现。
医疗质量检测技术展|你真的了解高压蒸汽灭菌器吗?
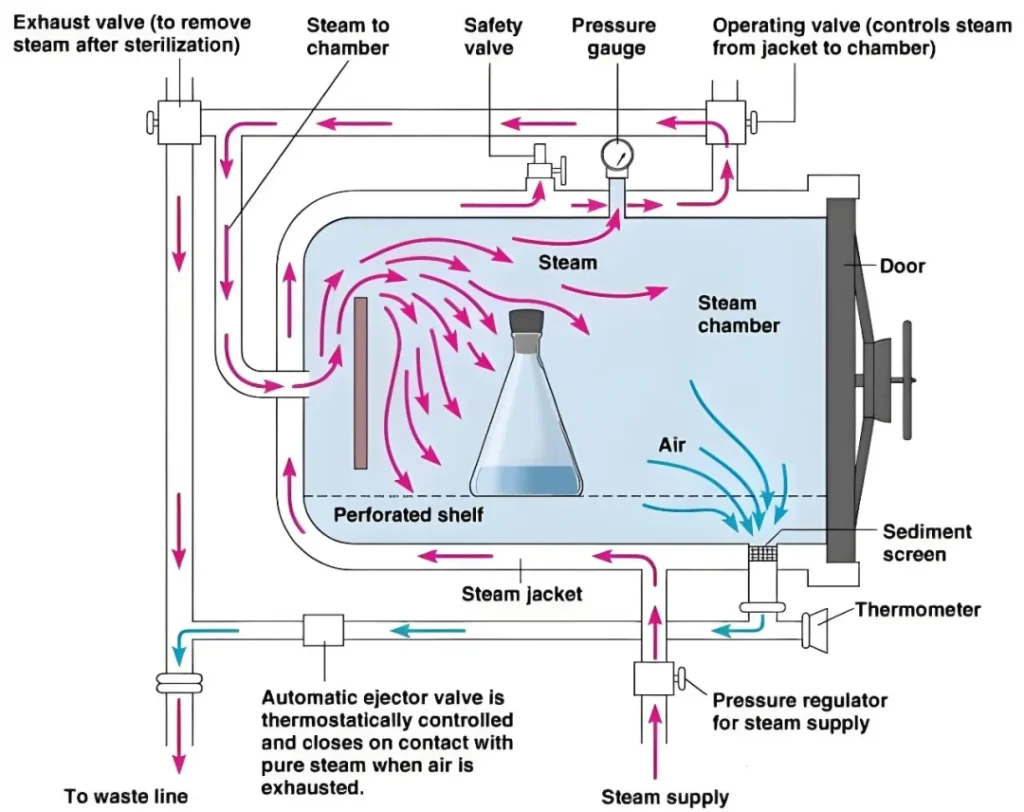
高压灭菌器是一种常用的灭菌设备,采用电热丝加热。当水加热到一定温度时,水就会变成蒸汽。
上海医疗检测设备展l 除菌过滤器的压力监测

《新版GMP附录1:无菌药品》中无菌药品的定义为:无菌药品是指法定药品标准中列有无菌检查项目的制剂和原料药,一般包括注射剂、无菌原料药、眼用制剂、无菌软膏剂、无菌混悬剂等。药品的无菌保证取决于合理且经过验证的灭菌工艺过程、良好的无菌保证体系以及生产过程中严格的GMP管理。
医疗质量检测技术展|ECA:关于欧盟无菌附录1的问答

是否有一个正式流程来确定附录1需要应用到API制造的范围?它在API制造期间如何被应用,以及如何系统地做出这些决定?
上海医疗检测设备展|洁净区的压差控制
洁净室与周围的空间必须维持一定的压差,并应按工艺要求决定维持正压差,通常情况洁净度高的区域流向洁净度低的区域,使洁净室的洁净度不受到污染空气的干扰。
医疗质量检测技术展|收藏!质量体系增加9001后,质量手册调整内容

医疗器械生产监管要求企业建立适应的质量体系以确保产品生产质量可控,但很多企业建立时仅纳入了GMP和13485,后续在做三方认证时意识到了9001的需求,故在原有体系上进行增加9001,这里就质量手册的改动部分做分享