



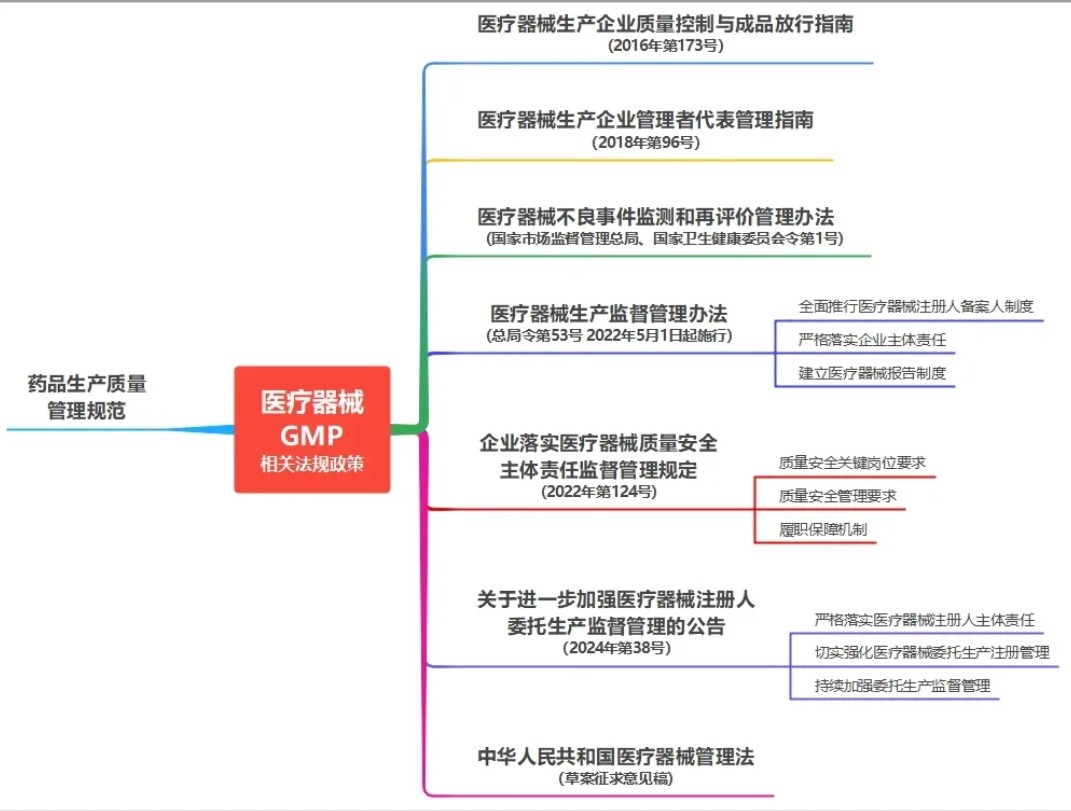
由医疗质量检测技术及测试仪器展知悉,医疗器械GMP及其附录对于规范行业生产质量管理起到了非常重要的指导作用。然而,随着行业的发展以及监管思路的优化升级,关于医疗器械GMP修订的消息不时会发出。
比如,早在2022年12月发布的《企业落实医疗器械质量安全主体责任监督管理规定》解读就提到,正在组织修订中的医疗器械生产质量管理规范将与《规定》衔接。

图片来源:炮打质量部
而本次所谓的小道消息是从飞行检查老师那里打探到的,随着《医疗器械管理法》(征求意见稿)的发布,相信医疗器械GMP升级落地应该不会太久了。那么,升级会从哪些方面入手呢?透过近些年相关法规政策的更新和完善,大概能够看出一些端倪,简单推测梳理如下,仅供参考。
图片来源:炮打质量部
企业落实医疗器械质量安全主体责任
我国的医疗器械行业经历了40多年的发展,医疗器械生产企业均已建立质量管理体系,风险管理理念被广泛接受和践行,逐步走向相对成熟的阶段。
而成熟的第一个特征就是责任担当,企业而非药监局才是医疗器械质量安全的第一责任人,所以,新修订的医疗器械GMP应该会衔接《企业落实医疗器械质量安全主体责任监督管理规定》进行相应的完善。
那么,《规定》主要内容是什么?
主要包括三方面内容:
一是质量安全关键岗位要求,明确生产企业质量安全关键岗位人员包括企业法定代表人和主要负责人、管理者代表、质量管理部门负责人,细化各岗位职责和任职条件。
二是质量安全管理要求,规定了质量安全管理调度和风险会商制度,细化委托生产管理、产品放行等关键环节管理要求,明确各环节负责人员。
三是履职保障机制,要求企业制定质量安全关键岗位说明书并对相关人员进行岗前培训和继续教育,规定生产企业管理者代表、质量管理部门负责人应当在职在岗,明确尽职免责制度和企业对相关人员的奖惩制度。
管理者代表
医疗器械GMP关于管理者代表的要求如下:
企业负责人应当确定一名管理者代表。
管理者代表应当负责建立、实施并保持质量管理体系,报告质量管理体系的运行情况和改进需求,提高员工满足法规、规章和顾客要求的意识。
而药监局2018年针对管理者代表的指南专门发布了《医疗器械生产企业管理者代表管理指南》(2018年第96号),对管代的职责有10条要求,且《企业落实医疗器械质量安全主体责任监督管理规定》解读也提到管代指南将会适时修订。
更有地方的药监局如山东前段时间已经开始征求《医疗器械生产企业管理者代表考评细则》的通告。
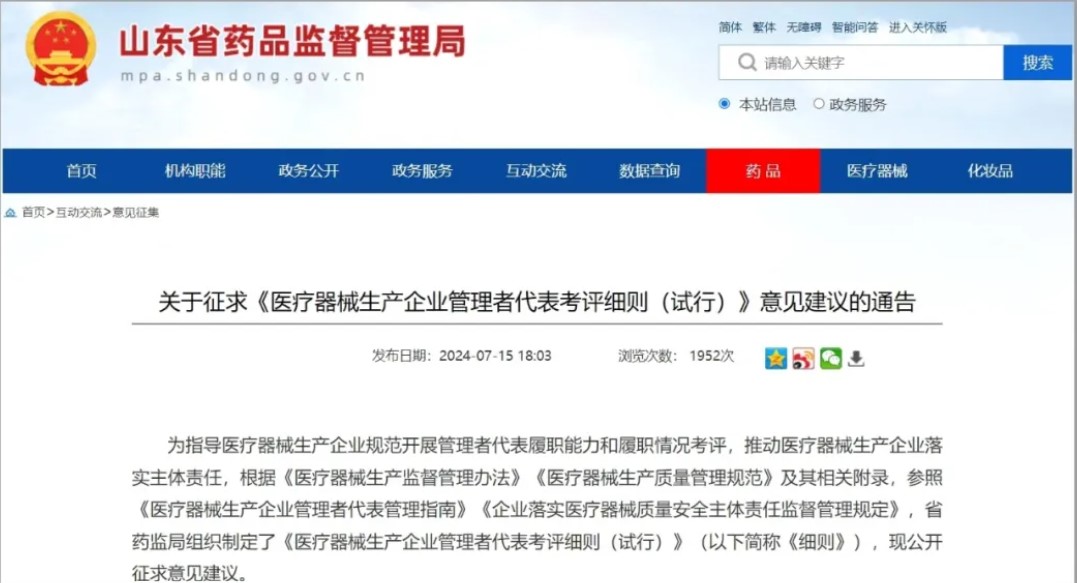
由此可见,医疗器械GMP对于管理者代表相关的要求可能会细化和完善。
医疗器械注册人委托生产

2021版《医疗器械监督管理条例》(国务院令 第739号)最大的调整莫过于注册人制度,其中涉及委托生产相关的生产质量管理,医疗器械GMP未能针对性地涵盖,所以不难推测,新修订的GMP将会完善委托生产方面的规范。
大概内容可以通过《关于进一步加强医疗器械注册人委托生产监督管理的公告(2024年第38号)》来了解。
再联想到《医疗器械管理法》(征求意见稿)关于“注册证可以转让”的提法,相关转让注册证产品的生产质量管理大概率也会纳入。
不良事件监测和再评价管理
作为国家市场监督管理总局改组后的第1号令,2018年8月13日国家市场监督管理总局、国家卫生健康委员会令第1号公布的《医疗器械不良事件监测和再评价管理办法》体现监管侧重点后置的意志,也更加完善全生命周期的监督管理。

而《医疗器械管理法》(征求意见稿)则将原来的“不良事件监测制度”调整为“警戒制度”,进一步扩大了各种有害事件的监测范围。
“第一百零七条 国家建立医疗器械警戒制度,对医疗器械不良事件以及其他与使用医疗器械有关的导致或者可能导致人体伤害的各种有害事件进行监测、识别、评估和控制。”
对应的,医疗器械GMP不良事件相应板块应该会改为警戒监测。
落实报告制度

医疗器械生产监督管理办法(总局令第53号 2022年5月1日起施行)要求落实报告制度。
那么,医疗器械注册人、备案人、受托生产企业应该如何落实报告制度?
为进一步落实医疗器械注册人、备案人、受托生产企业主体责任,《办法》建立医疗器械报告制度,并规定相应法律责任。医疗器械注册人、备案人、受托生产企业应当按照规定执行医疗器械报告制度。
一是落实自查报告制度。医疗器械注册人、备案人、受托生产企业应当每年对质量管理体系的运行情况进行自查,按照医疗器械生产质量管理体系年度自查报告编写指南要求编写自查报告,并于次年3月31日前向所在地药品监督管理部门提交自查报告。进口医疗器械注册人、备案人由其代理人向代理人所在地省、自治区、直辖市药品监督管理部门提交自查报告。
二是落实生产产品品种报告制度。医疗器械生产企业应当向药品监督管理部门报告所生产的产品品种情况。增加生产产品品种的,应当向原生产许可或者生产备案部门报告,涉及委托生产的,还应当提供委托方、受托生产产品、受托期限等信息。医疗器械生产企业增加生产产品涉及生产条件变化,可能影响产品安全、有效的,应当在增加生产产品30个工作日前向原生产许可部门报告,原生产许可部门应当及时开展现场核查。属于许可事项变化的,应当按照规定办理相关许可变更。
三是落实生产条件变化报告制度。医疗器械注册人、备案人、受托生产企业的生产条件发生变化,不再符合医疗器械质量管理体系要求的,应当立即采取整改措施;可能影响医疗器械安全、有效的,应当立即停止生产活动,并向原生产许可或者生产备案部门报告。受托生产企业应当及时将变化情况告知医疗器械注册人、备案人。
四是落实重新生产报告制度。医疗器械生产企业连续停产一年以上且无同类产品在产的,重新生产时,应当进行必要的验证和确认,并书面报告药品监督管理部门。可能影响质量安全的,药品监督管理部门可以根据需要组织核查。
所以小编猜测后续新版医疗器械GMP会呼应体现报告制度相应要求。
从质量控制到质量保证

熟悉质量管理发展历史的小伙伴应该都有印象,质量管理已经从最初的质量控制(从已生产的产品里挑选不良品)到质量保证(预防不良的发生),进一步发展为全面质量管理阶段(从组织层面而非单纯产品层面提升质量)。
而我们的医疗器械GMP目前还停留在第一阶段——质量控制阶段,还要很大的提升空间。

对照一下隔壁药品的管理就会发现,早在2011年施行的药品GMP已经采用质量保证、质量控制和质量风险管理的结构进行质量管理的表述。
不难推测,新的医疗器械GMP大概率会向药品和时代看齐,完善质量管理的结构,纳入质量保证部分。

图片来源:炮打质量部
综上所述,大胆推测新版的医疗器械GMP可能会从落实企业主体责任、完善管理者代表管理、纳入注册人委托生产质量管理、将不良事件监测扩大为警戒监测、落实报告制度和从质量控制到质量保证等方面进行升级修订。限于阅历和能力,仅期望通过本次梳理给各位小伙伴一些启发,为进一步提升企业质量管理水平提供一些思路。不正之处,欢迎拍砖讨论。
想要了解更多请前往医疗质量检测技术及测试仪器展
文章来源:炮打质量部
若涉及侵权,请立刻联系删除
关键字:


