



医疗质量检测技术展|医疗器械注册质量管理体系核查指南介绍

本指南旨在加强医疗器械注册质量管理体系的核查管理,确保核查工作的质量。依据包括《医疗器械监督管理条例》、《医疗器械注册与备案管理办法》、《体外诊断试剂注册与备案管理办法》、《医疗器械生产监督管理办法》、《医疗器械生产质量管理规范》、《医疗器械临床试验质量管理规范》及《医疗器械注册自检管理规定》等法规。
上海医疗测试仪器展|ISO13485医疗器械质量管理标准 问与答 (一)

医疗器械质量管理体系起源于2000年我国发布的第22号医疗器械质量管理体系核查要求,并在此基础上历经九年发展形成了我国第一版医疗器械生产质量管理规范。随着医疗器械行业的快速发展,我国于2014年对GMP进行了修正并形成了最新的标准规范。同时,其他国家和地区也陆续建立了相应的质量管理体系标准法规,例如美国的CDMP及后续QSF20法规,欧盟的ENISO13485标准及其多次修订等。在70年代,美国率先制定了CDMP法规,并在其基础上于1996年发布针对质量管理体系要求的QSF20法规。欧盟则从1993年开始酝酿并报告了EN1N46001标准法规,但后来对该标准进行了修订和完善。国际标准化组织在1996年发布了基于ISO9001系列的两个医疗器械质量管理体系标准,即ISO13485:1996版与ISO13481:1996版,并随9001版本升级不断调整优化,最终在2016年发布了全新的ISO13485:2016版标准。
医疗质量检测技术及测试仪器展|医疗器械GMP对“空调净化系统”的要求

医疗器械的生产过程和加工工序必须在洁净室(区)内进行,并达到规定的洁净度级别要求,不同医疗器械适用的洁净度级别要求不一致。对洁净室(区)的工作环境必须严格控制,才能有效地防止工作环境对医疗器械的污染,保证产品质量和使用者安全。而洁净度级别的控制主要通过空调净化系统的科学合理选择、设计、维护实现。
医疗质量检测技术展|医械生物相容性评价的目标和方法

根据ISO 10993-1:2018的定义,生物相容性是指医疗器械或材料在一个特定应用中引起恰当宿主反应的能力。“生物相容”与“生物不相容”不是某种材料天然的或绝对的“标签”,而是需要结合材料的具体性能和特定的(临床)应用场景进行判断。同时,生物相容性是一个动态概念,植入物植入人体后会对特定生物组织环境产生物理和化学影响,引起生物学反应;反之,生物组织也会对植入物产生影响,使之发生物理或化学变化,两者的相互作用会一直持续。即使植入物被完全去除或被人体完全吸收,其影响还将持续一段时间。
上海医疗检测设备展|医疗器械认证过程中的风险管理要点

当我们谈论医疗器械行业的风险管理时,有些人可能会认为这是一个令人生畏的混乱过程。一开始可能会让人不知所措,但这对确保我们开发的器械在整个生命周期内安全有效至关重要。这不仅仅是为了满足监管要求—虽然这绝对是其中的一部分—而是为了做出明智的决策,保护患者并降低器械失效的可能性。
医疗质量检测技术及测试仪器展|质量管理体系构建中,值得避坑的案例学习!

近期国家药监局在对部分医疗器械企业进行飞检过程中,发现部分企业的质量管理体系存在较大缺陷,本次笔者就针对飞检的2家企业的问题进行收集整理,希望对各企业在质量管理体系的管理中有所借鉴和帮助。
医疗质量检测技术展|质量部DQE、SQE、CQE、PQE 岗位职责有哪些?
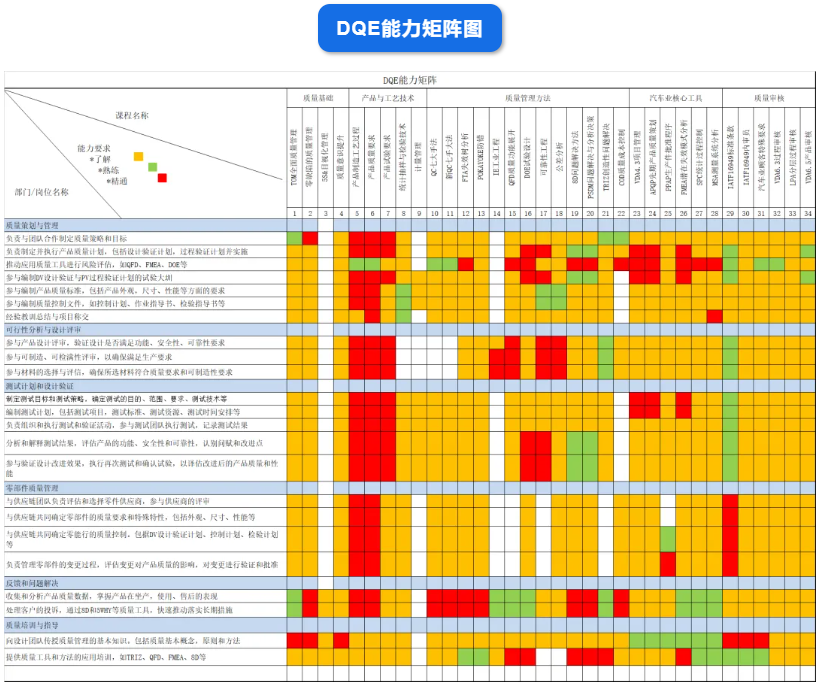
DQE是在产品设计和开发过程中负责质量管理和质量保证的专业人员。他们的主要职责是确保产品设计符合质量标准和客户要求。以下是DQE设计质量工程师的常见工作职责:
1. 质量计划和策略:制定和执行产品设计和开发阶段的质量计划和策略,确保产品符合质量标准和规范。
医疗器械行业展览|原子层沉积(ALD)揭秘:成功开发、优化和表征 ALD 的 10 个步骤
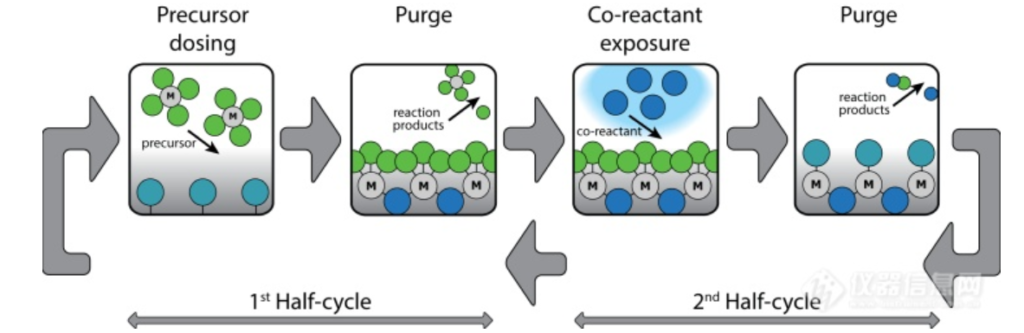
原子层沉积技术(ALD)是一种一层一层原子级生长的薄膜制备技术。理想的 ALD 生长过程,通过选择性把不同的前驱体暴露于基片的表面交替,在表面化学吸附并反应形成沉积薄膜。
医疗器械行业展览丨医疗器械摸底测试

医疗器械摸底测试是在医疗器械开发的早期阶段进行的初步测试,旨在了解医疗器械的基本性能和特征,初步评估其适应性和可行性,防止在医疗器械注册检验过程中耽误更多的时间,延缓上市周期。
医疗器械制造展丨设计变更的质量管理实践

设计变更是一个关键的医疗器械质量管理体系中的环节,需要我们采取适当的步骤,确保变更的安全性、有效性以及符合法规要求。结合以往实战经验,提供以下是一些实施建议,期望可以帮助我们在设计变更活动中做得更好: