



医疗质量检测技术及测试仪器展|读懂GB9706医疗器械强制标准,离不开这5大问题!

GB9706.1-2020《医用电气设备 第1部分:基本安全和基本性能的通用要求》已经从2023年5月1日起开始实施。那么,企业在执行GB9706系列标准时经常遇到哪些问题?久顺收集汇总不少企业相关询问,本期一并解答。
医疗质量检测技术及测试仪器展|医疗器械GMP:检验设备是否都需要校验

企业生产医疗器械时,为确认产品性能指标以及生产和检验环境的洁净度,需要对产品性能指标、洁净区环境、工艺用水、产品无菌性能、环氧乙烷残留量(EO灭菌)等进行检验。质量检验的结果是否准确可靠,取决于检验设备测量的结果的准确性,企业在使用检验设备时,需要提供客观证据,证明设备符合要求。
医疗质量检测技术展|医疗器械注册检验常见问题答疑汇总

A:维氏硬度检测不需要提供单独的试样,在送检样品上取样测试;洛氏硬度检测若被测部位形状不规则时,无法直接对样品进行测试,应提供原材料试样块,并提供材质、热处理工艺一致性声明。
上海医疗测试仪器展|医疗器械同品种临床评价报告怎么写?
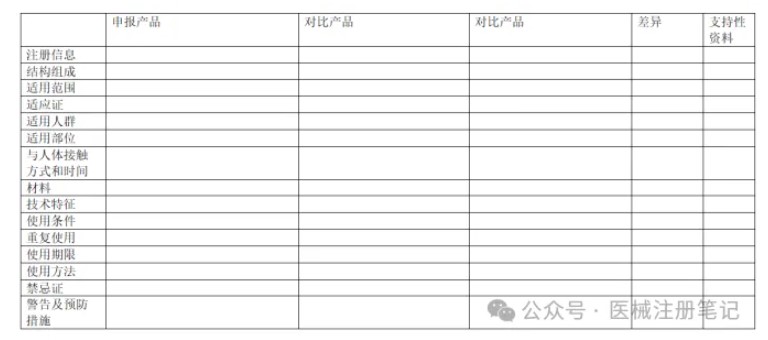
同品种比对是医疗器械注册申报过程中进行临床评价的重要方式之一,包括通过等同器械的临床数据进行临床评价以及使用可比器械的临床数据进行部分临床评价。同品种比对是企业节省大量研发费用,缩短新产品上市时间的优先选择。
医疗质量检测技术及测试仪器展|无源医疗器械安全性评价第二部分:货架有效期
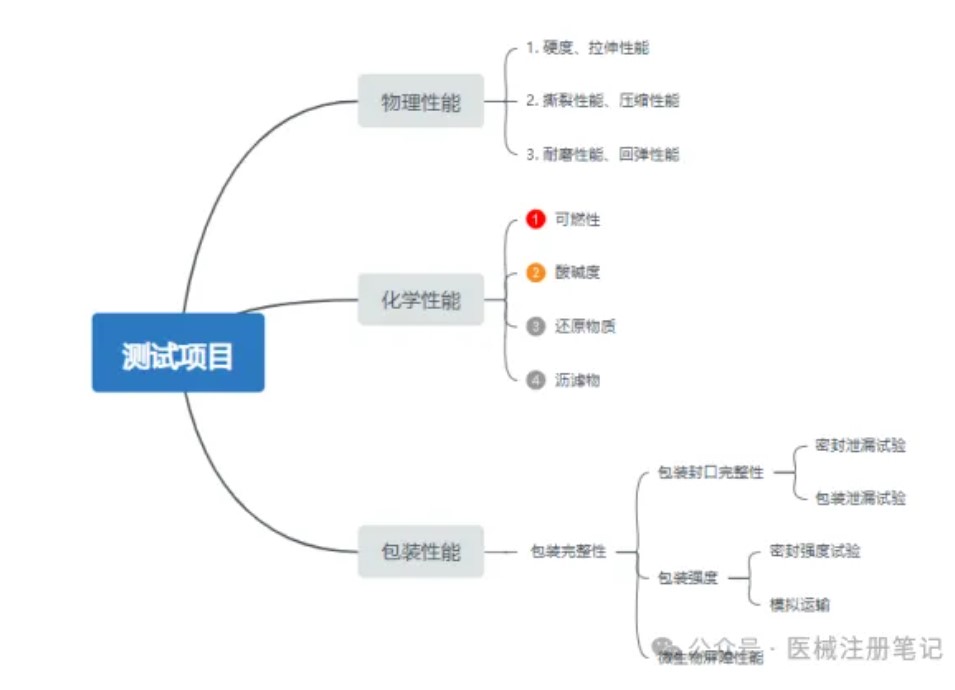
医疗器械注册阶段,货架有效期部分研究至关重要,货架有效期研究是确保产品在规定期限内能够正常发挥预期功能的重要证据。当医疗器械超过货架有效期研究规定的最大期限,器械的性能可能无法得到保证,在使用时则存在潜在风险。
上海医疗检测设备展|一文了解医疗器械发光测定仪是什么

高灵敏度:化学发光测定技术具有很高的灵敏度,能够检测到低浓度的生物分子,适用于早期疾病的诊断。
医疗质量检测技术展|医疗器械“体考”到底是什么?

《医疗器械生产监督管理办法》明确规定:医疗器械生产企业应当按照医疗器械生产质量管理规范的要求,建立质量管理体系并保持有效运行。申请人在申请产品注册时,受理注册申请的药品监督管理部门在产品技术审评时,认为有必要对质量管理体系进行核查的,会组织开展质量管理体系核查。
医疗质量检测技术展|医疗器械注册质量管理体系核查指南介绍

本指南旨在加强医疗器械注册质量管理体系的核查管理,确保核查工作的质量。依据包括《医疗器械监督管理条例》、《医疗器械注册与备案管理办法》、《体外诊断试剂注册与备案管理办法》、《医疗器械生产监督管理办法》、《医疗器械生产质量管理规范》、《医疗器械临床试验质量管理规范》及《医疗器械注册自检管理规定》等法规。
上海医疗测试仪器展|ISO13485医疗器械质量管理标准 问与答 (一)

医疗器械质量管理体系起源于2000年我国发布的第22号医疗器械质量管理体系核查要求,并在此基础上历经九年发展形成了我国第一版医疗器械生产质量管理规范。随着医疗器械行业的快速发展,我国于2014年对GMP进行了修正并形成了最新的标准规范。同时,其他国家和地区也陆续建立了相应的质量管理体系标准法规,例如美国的CDMP及后续QSF20法规,欧盟的ENISO13485标准及其多次修订等。在70年代,美国率先制定了CDMP法规,并在其基础上于1996年发布针对质量管理体系要求的QSF20法规。欧盟则从1993年开始酝酿并报告了EN1N46001标准法规,但后来对该标准进行了修订和完善。国际标准化组织在1996年发布了基于ISO9001系列的两个医疗器械质量管理体系标准,即ISO13485:1996版与ISO13481:1996版,并随9001版本升级不断调整优化,最终在2016年发布了全新的ISO13485:2016版标准。
上海医疗测试仪器展|医疗器械生物学评价流程与思路
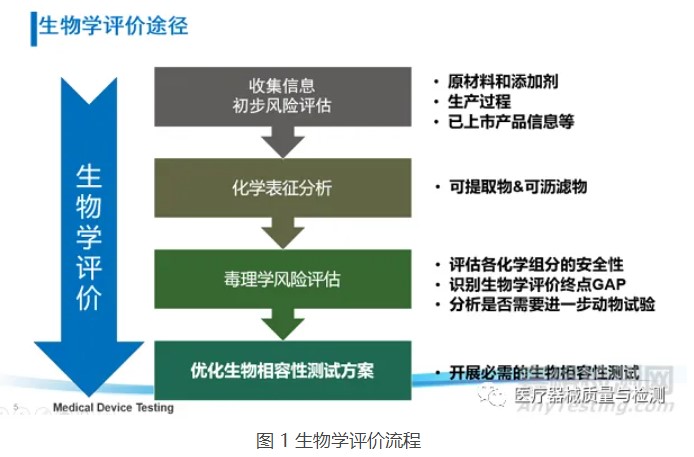
考虑器械潜在的生物学风险,并不意味着要针对所有风险点都开展生物学测试,还可通过“评价”的方式开展物理/化学表征及毒理学风险评估、基于已有的临床应用历史和人体接触数据对器械的生物学风险进行评估。在近年来更新的ISO 10993-1:2018和FDA年发布的关于ISO 10993-1的应用指南中,均强调了通过化学表征测试和毒理学风险评估(ISO 10993-18和ISO 10993-17)进行生物学评价的思路,不仅可以豁免不必要的生物相容性测试及避免人力、物力和动物资源的浪费,还可以基于已有的研究数据更加充分地评估器械(尤其是持久性植入的高风险类器械)中潜在的生物安全性风险(如慢性毒性、致癌性和生殖毒性等),从而优化生物学测试方案,最终达到器械安全性评价的目的。