



医疗质量检测技术展|医疗器械必学法规:医疗器械使用质量监督管理办法(国家食药监管理总局令第18号)

这份法规文件主要规定了医疗器械在使用环节的质量管理要求和监督管理措施,旨在确保医疗器械的使用安全、有效。以下是文件中的关键知识点:
医疗质量检测技术及测试仪器展|医疗器械独立软件核查中对相关标准的思考
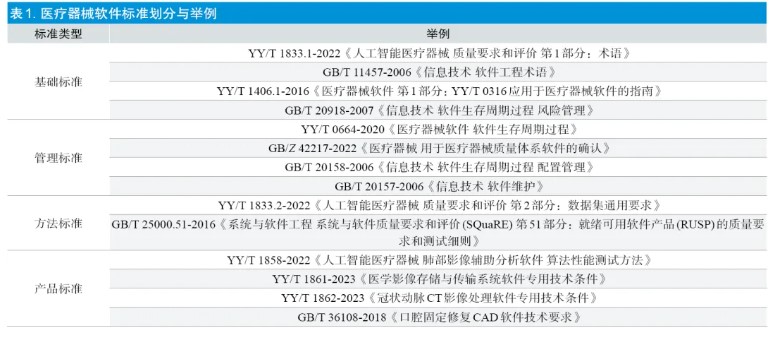
随着计算机技术的进步,医疗器械软件行业正处于高速发展期。近年来,上海市独立软件产品申报数量大幅增长。相较于其他医疗器械产品,软件产品的多样化程度高、技术发展迅速、产品迭代更新快、非实体导致缺陷隐蔽性强,因此产品的安全有效性评价难度较高。在审评核查的过程中发现,医疗器械软件产品常见基本概念不清晰、性能指标和检验方法不明确、产品验证与确认不充分、生存周期过程控制不足等问题。标准在规范产品性能指标、保证产品安全有效性、促进创新和技术进步等方面发挥着重要的作用。随着医疗器械软件行业的迅速发展以及产品自身的复杂性,对配套标准也提出了更高的要求。本文在概要论述医疗器械软件标准发展现状的基础上,分析了目前在医疗器械独立软件审评核查中与标准相关的常见问题,探讨了如何更加有效地开展医疗器械软件质量评价工作,并提出了软件标准未来发展的若干思考和建议,以期加强标准的指导意义、统一评价尺度、促进医疗器械软件的标准化进程。
医疗质量检测技术及测试仪器展|医疗器械GMP要升级

2014年12月,原国家食品药品监督管理总局(CFDA)发布了医疗器械生产质量管理规范(以下简称医疗器械GMP)(2014年第64号),并随后陆续推出了无菌医疗器械、植入性医疗器械、体外诊断试剂、定制式义齿和独立软件(注:NMPA发布)等5个附录。
医疗质量检测技术展|医疗器械软件注册检验要点及方法研究
目的:针对医疗器械软件注册检验要点和检测方法进行研究,为加强医疗器械软件的质量保证工作提供参考。方法:以医疗器械软件注册指导原则为基础,梳理不同医疗器械软件的特点和分类,明确软件产品技术要求的编制指南,并且结合通用要求、质量要求、专用要求、安全要求四个方面的要求,对注册检验内容进行解读,最后总结常用检测方法和检测工具。结果:梳理的医疗器械软件注册检验要点可以为注册申请人编写产品技术要求提供指导,总结的检验方法和检测工具可以给检测机构开展相关评测提供借鉴。结论:医疗器械软件检验应从通用要求、质量要求、专用要求和安全要求四个方面进行,检测主要涉及功能测试和性能测试两大类,性能测试通常需借助专业工具进行。
医疗质量检测技术展|人工智能医疗器械注册审查基本原则
人工智能算法的类型不同,其算法特性、适用场景也不同,评价重点亦有所侧重;同时,不同类型的人工智能算法可组合使用,需结合各算法特性和算法组合形式进行整体评价。
医疗质量检测技术及测试仪器展|设计技术考量点
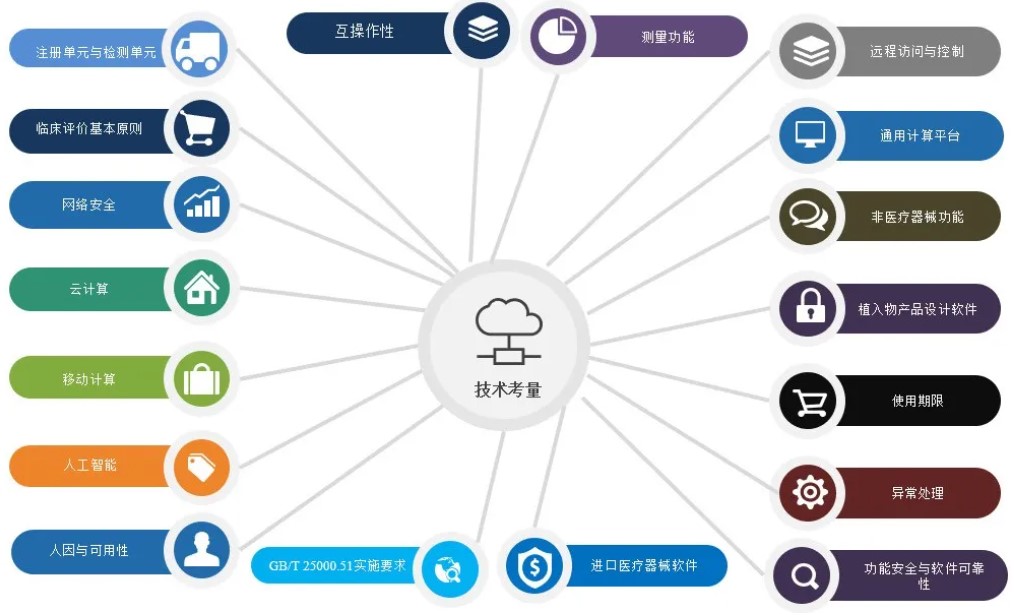
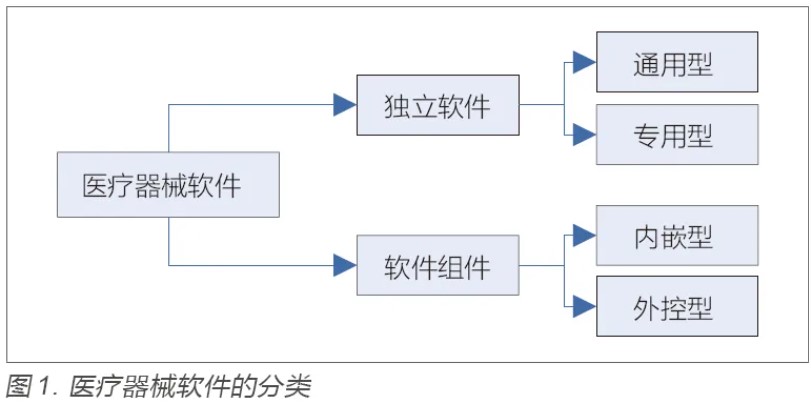
在进行医疗期器械软件设计过程中,我们可以从以下18点来进行技术考量。分别为1.注册单元与检测单元、2.临床评价基本原则、3.网络安全、4.云计算、5.移动计算、6.人工智能、7.人因与可用性、8.互操作性、9.测量功能、10.远程访问与控制、11.通用计算平台、12.非医疗器械功能、13.植入产品设计软件、14.使用期限、15.异常处理、16.功能安全与软件可靠性、17.GB/T25000.51实施要求和18.进口医疗器械软件等要求。
医疗质量检测技术展|医疗器械原材料变更的基本流程

医疗器械的预期用途、技术要求、安全有效性、有效期等等,很大程度上取决于原材料。
产品注册时确定下来的原材料,轻易不能变化。
但是,企业在实际经营过程中,常常为了降低成本、改善某方面性能、完善供应链、符合升版的标准,或主动、或被动地对原材料进行变更。
医疗质量检测技术及测试仪器展|医疗器械生物相容性简介、流程、路径和数据
医疗器械生物相容性评价是确保医疗器械安全性和有效性的重要环节,它涉及到对医疗器械在与人体接触时可能产生的生物学反应和风险的全面评估。根据国家标准GB/T 16886.1-2022《医疗器械生物学评价 第1部分:风险管理过程中的评价与试验》,生物相容性评价应作为医疗器械总体评价和开发过程的一部分,贯穿于产品的全生命周期。
医疗质量检测技术展|医疗器械生产组织的质量管理体系需满足的四方面要求

医疗器械生产组织的质量管理体系需满足四方面要求,即质量管理体系GB/T42061标准要求、法规要求、顾客要求和组织自身要求。
医疗质量检测技术及测试仪器展|读懂GB9706医疗器械强制标准,离不开这5大问题!

GB9706.1-2020《医用电气设备 第1部分:基本安全和基本性能的通用要求》已经从2023年5月1日起开始实施。那么,企业在执行GB9706系列标准时经常遇到哪些问题?久顺收集汇总不少企业相关询问,本期一并解答。