



医疗质量检测技术展|医疗器械软件注册检验要点及方法研究
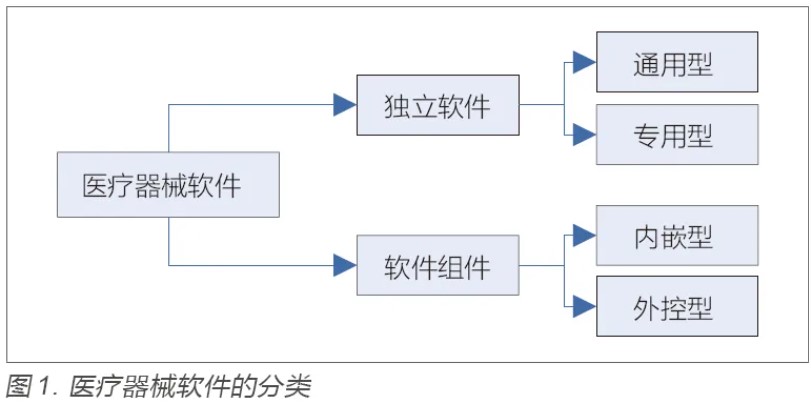
目的:针对医疗器械软件注册检验要点和检测方法进行研究,为加强医疗器械软件的质量保证工作提供参考。方法:以医疗器械软件注册指导原则为基础,梳理不同医疗器械软件的特点和分类,明确软件产品技术要求的编制指南,并且结合通用要求、质量要求、专用要求、安全要求四个方面的要求,对注册检验内容进行解读,最后总结常用检测方法和检测工具。结果:梳理的医疗器械软件注册检验要点可以为注册申请人编写产品技术要求提供指导,总结的检验方法和检测工具可以给检测机构开展相关评测提供借鉴。结论:医疗器械软件检验应从通用要求、质量要求、专用要求和安全要求四个方面进行,检测主要涉及功能测试和性能测试两大类,性能测试通常需借助专业工具进行。
医疗质量检测技术及测试仪器展|US FDA关于无菌器械灭菌的要求

无菌类器械510(k)中,成熟的灭菌方法包括蒸汽灭菌、干热灭菌、环氧乙烷(EO)灭菌、辐射灭菌、汽化过氧化氢、臭氧等。正在开发的全新灭菌技术并将用于I类和II类器械生产的,被视为新方法。对于采用新灭菌技术灭菌的器械,如果操作不当,会带来灭菌不到位的重大风险。因此,应密切评估使用这些技术灭菌的器械是否符合GMP。FDA计划先审查其制造机构再授予510(k)许可,这将有助于确保器械的安全性和有效性,并降低对人类健康的风险。
医疗质量检测技术展|三类降级为二类,FDA为什么那么大的决心?
FDA对Class III检测的要求是每个经过上市前批准程序的器械都有责任独立提供数据和信息,以合理保证安全性和有效性,光是General Control和Special Control都还不够。整个过程十分漫长,涉及大量的临床试验和数据收集。FDA只有在确定足够的科学证据支持该设备对其预期用途安全有效后才会批准申请。
医疗质量检测技术展|人工智能医疗器械注册审查基本原则
人工智能算法的类型不同,其算法特性、适用场景也不同,评价重点亦有所侧重;同时,不同类型的人工智能算法可组合使用,需结合各算法特性和算法组合形式进行整体评价。
医疗质量检测技术及测试仪器展|网络安全更新
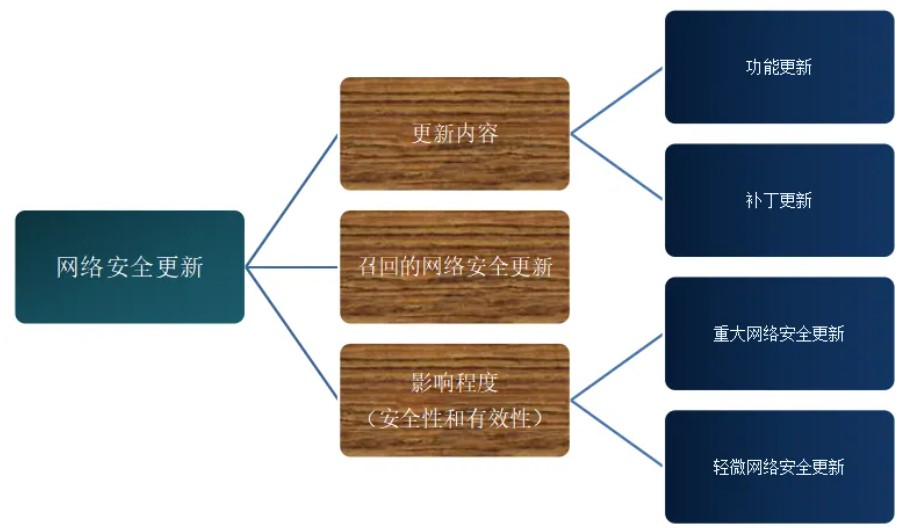
医疗器械网络安全更新从内容上可分为功能更新、补丁更新,类似于增强类软件更新、纠正类软件更新。
根据其对医疗器械安全性和有效性的影响程度分为以下两类:
医疗质量检测技术及测试仪器展|设计技术考量点
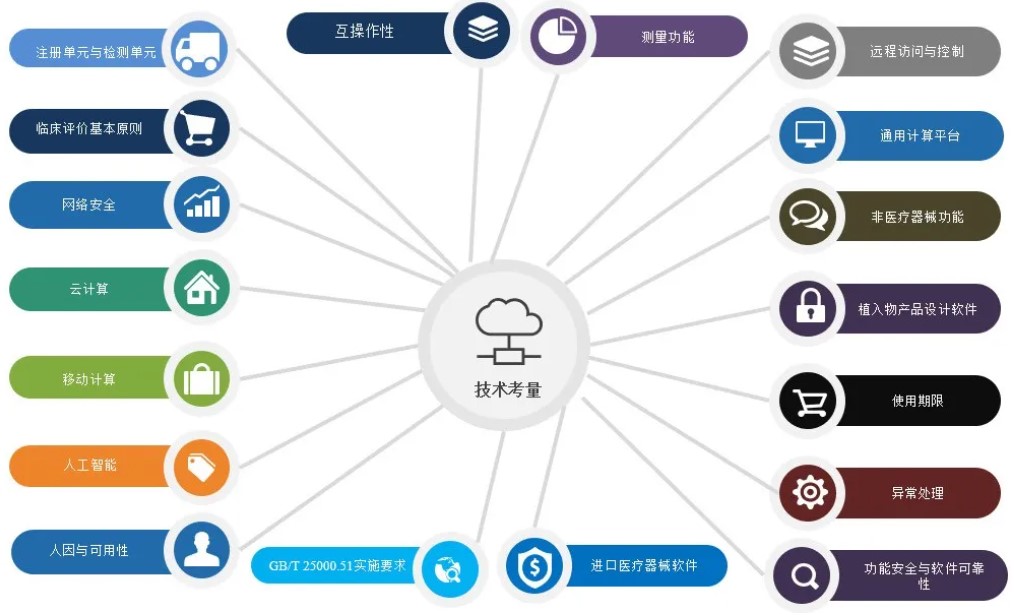
在进行医疗期器械软件设计过程中,我们可以从以下18点来进行技术考量。分别为1.注册单元与检测单元、2.临床评价基本原则、3.网络安全、4.云计算、5.移动计算、6.人工智能、7.人因与可用性、8.互操作性、9.测量功能、10.远程访问与控制、11.通用计算平台、12.非医疗器械功能、13.植入产品设计软件、14.使用期限、15.异常处理、16.功能安全与软件可靠性、17.GB/T25000.51实施要求和18.进口医疗器械软件等要求。
医疗质量检测技术及测试仪器展|再谈MDR和FDA的器械可用性测试
测试完成后,您将需要制作一份可用性工程报告,该报告需要提交给您的医疗设备销售市场的相应监管机构。需要注意的是,这份报告不应该简单地总结你的可用性验证测试,而是需要一个非常具体的格式。
医疗质量检测技术展|医疗器械原材料变更的基本流程

医疗器械的预期用途、技术要求、安全有效性、有效期等等,很大程度上取决于原材料。
产品注册时确定下来的原材料,轻易不能变化。
但是,企业在实际经营过程中,常常为了降低成本、改善某方面性能、完善供应链、符合升版的标准,或主动、或被动地对原材料进行变更。
医疗质量检测技术及测试仪器展|医疗器械生物相容性简介、流程、路径和数据
医疗器械生物相容性评价是确保医疗器械安全性和有效性的重要环节,它涉及到对医疗器械在与人体接触时可能产生的生物学反应和风险的全面评估。根据国家标准GB/T 16886.1-2022《医疗器械生物学评价 第1部分:风险管理过程中的评价与试验》,生物相容性评价应作为医疗器械总体评价和开发过程的一部分,贯穿于产品的全生命周期。
医疗质量检测技术展|医疗器械生产组织的质量管理体系需满足的四方面要求

医疗器械生产组织的质量管理体系需满足四方面要求,即质量管理体系GB/T42061标准要求、法规要求、顾客要求和组织自身要求。