



上海医疗检测设备展|ISO13485和GB/T42061解读、导入与审核(条款7.3.3 设计和开发输入)
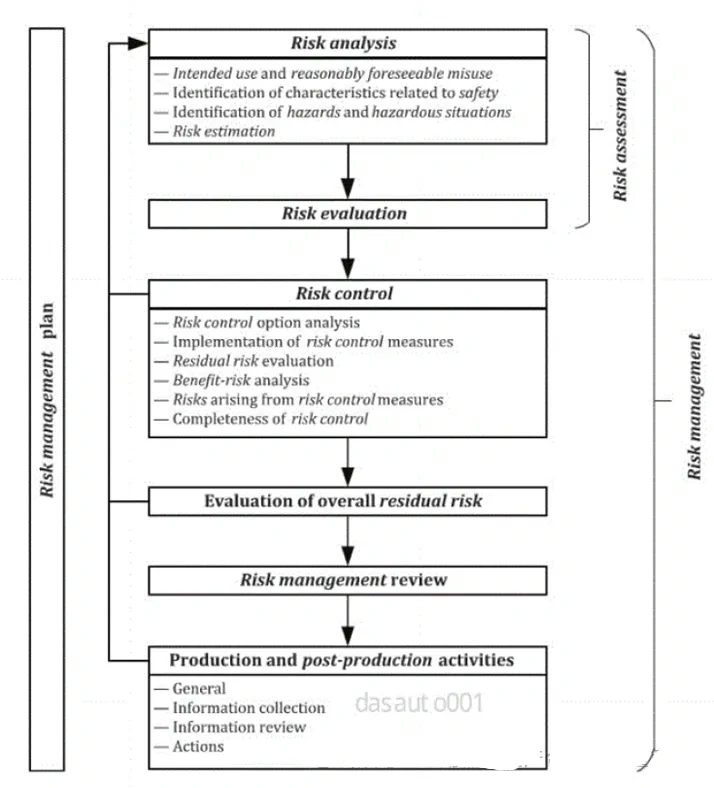
ISO13485:2016《医疗器械质量管理体系用于法规的要求》,由ISO国际标准化组织于2016年3月1 日正式发布,是一项应用于医疗器械领域的质量管理体系的国际标准,强调医疗器械的安全有效,组织提供的医疗器械要满足顾客和法规要求。
上海医疗测试仪器展|医疗器械测试项目清单:确保安全合规的关键步骤

在医疗器械行业,产品的安全性和合规性一直是至关重要的问题。在上海医疗测试仪器展上,这一点尤其受到关注。为了确保医疗器械在临床使用中能够安全可靠地发挥作用,医疗器械测试是必不可少的步骤。
医疗质量检测技术及测试仪器展|医疗器械EMC要求概览 1
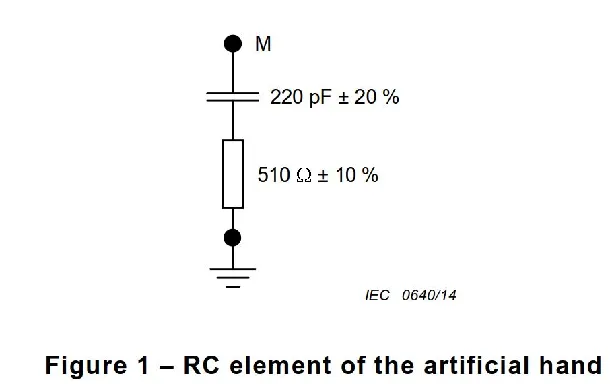
磁兼容性(EMC)是衡量电子设备在电磁环境中正常工作且不对该环境中任何事物构成不能承受的电磁骚扰的能力。对于医疗器械而言,由于其直接应用于人体,对EMC的要求更为严格。
医疗质量检测技术展|ECA:关于欧盟无菌附录1的问答

是否有一个正式流程来确定附录1需要应用到API制造的范围?它在API制造期间如何被应用,以及如何系统地做出这些决定?
上海医疗检测设备展|一文了解医疗器械检测的重要性和流程

医疗器械检测在保障患者安全、提升医疗质量以及推动医疗器械行业健康发展方面扮演着至关重要的角色。上海医疗检测设备 […]
医疗质量检测技术展|浅谈在用医疗器械质量检测的常见方法有哪些

在用医疗器械质量检测的常见方法多种多样,这些方法旨在确保医疗器械的安全性、有效性和符合相关标准。
上海医疗检测设备展|医疗器械检测流程详解:哪些项目不可忽略?

医疗器械检测是确保产品安全性和有效性的关键步骤。无论是在开发阶段还是在产品上市前,医疗器械检测都扮演着至关重要的角色。企业需要通过一系列严格的检测环节,才能确保产品满足各项法规要求,顺利通过监管机构的审批,进入市场。
上海医疗测试仪器展|欧盟与我国GMP无菌药品附录差异分析

无菌药品作为高风险产品,其在生产管理和质量管理上具有其高标准和特殊性。
上海医疗检测设备展|无菌医疗器械包装质量控制要点(下)

无菌包装生产技术已经成熟。随着法规和检测技术的不断完善,使用企业对质量要求在逐步提高。法规和质量标准的推出将进一步规范无菌包装产品的质量和性能,从而更好地保障无菌医疗器械的安全性和有效性。
医疗质量检测技术展|包装运输试验和环境试验

国家药监局发布的《医疗器械安全和性能基本原则》中明确规定:医疗器械的设计、生产和包装,包括申请人所提供的说明和信息,应确保在按照预期用途使用时,运输和贮存条件(例如:跌落、振动、温度和湿度的波动)不会对医疗器械的特性和性能,包括完整性和清洁度,造成不利影响。