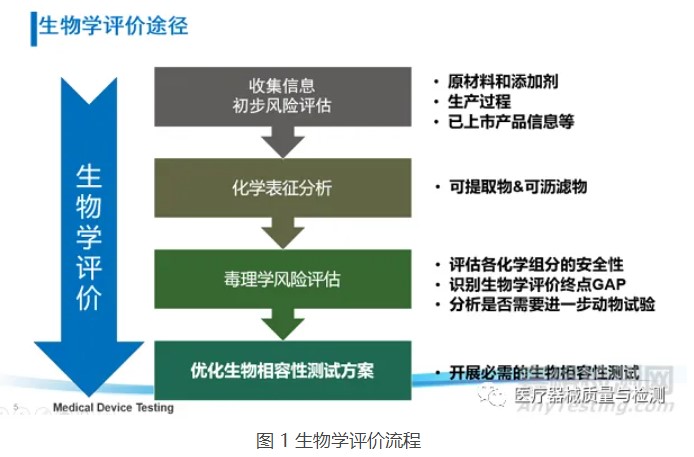
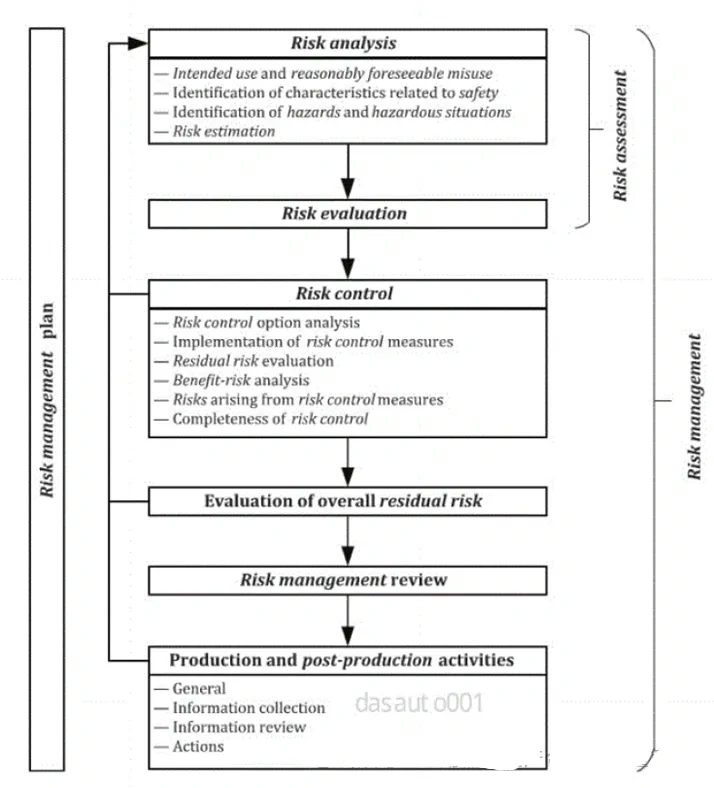
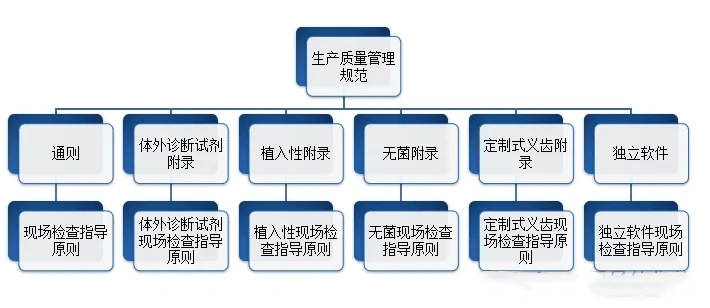
考虑器械潜在的生物学风险,并不意味着要针对所有风险点都开展生物学测试,还可通过“评价”的方式开展物理/化学表征及毒理学风险评估、基于已有的临床应用历史和人体接触数据对器械的生物学风险进行评估。在近年来更新的ISO 10993-1:2018和FDA年发布的关于ISO 10993-1的应用指南中,均强调了通过化学表征测试和毒理学风险评估(ISO 10993-18和ISO 10993-17)进行生物学评价的思路,不仅可以豁免不必要的生物相容性测试及避免人力、物力和动物资源的浪费,还可以基于已有的研究数据更加充分地评估器械(尤其是持久性植入的高风险类器械)中潜在的生物安全性风险(如慢性毒性、致癌性和生殖毒性等),从而优化生物学测试方案,最终达到器械安全性评价的目的。