EN|CN
首页 » 行业资讯
上海医疗检测设备展|医疗器械检测流程详解:哪些项目不可忽略?
上海医疗测试仪器展|欧盟与我国GMP无菌药品附录差异分析
上海医疗检测设备展|洁净区的压差控制
上海医疗检测设备展|无菌医疗器械包装质量控制要点(下)
医疗质量检测技术展|包装运输试验和环境试验
上海医疗检测设备展|医疗器械工艺用水验证
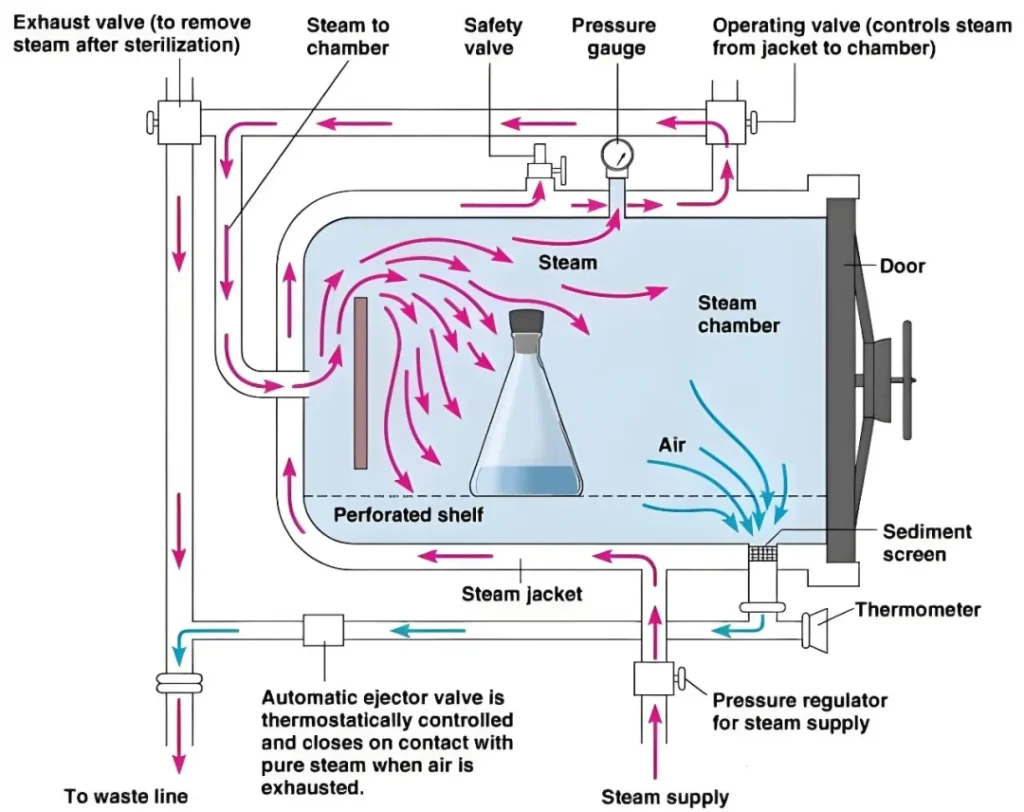
医疗质量检测技术展|你真的了解高压蒸汽灭菌器吗?
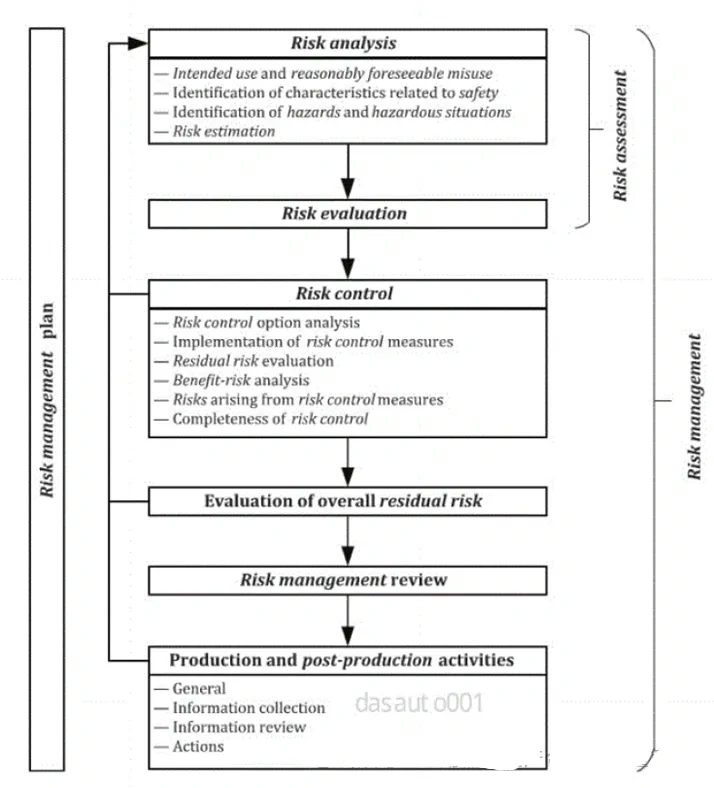
医疗质量检测技术及测试仪器展|验证与确认:医疗器械领域的深入解析与实例
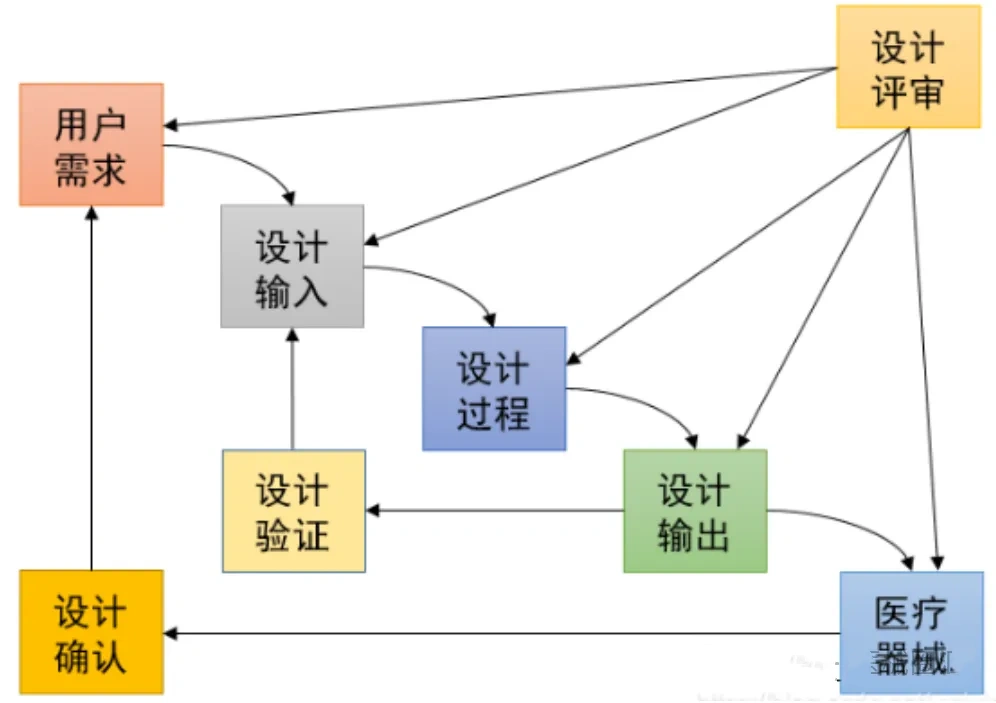
上海医疗检测设备展|ISO13485和GB/T42061解读、导入与审核(条款7.3.3 设计和开发输入)
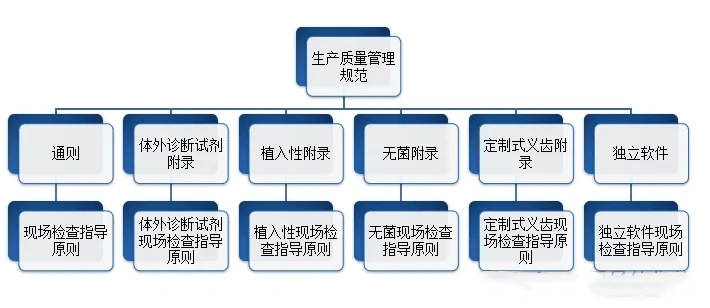
医疗质量检测技术展|中国GMP医疗器械生产质量管理规范包含哪些?