EN|CN
首页 » 行业资讯
医疗质量检测技术及测试仪器展|仪器校准标签有哪些常见错误?检查这几点就可以了
医疗质量检测技术展|一文读懂我国医疗器械唯一标识UDI!
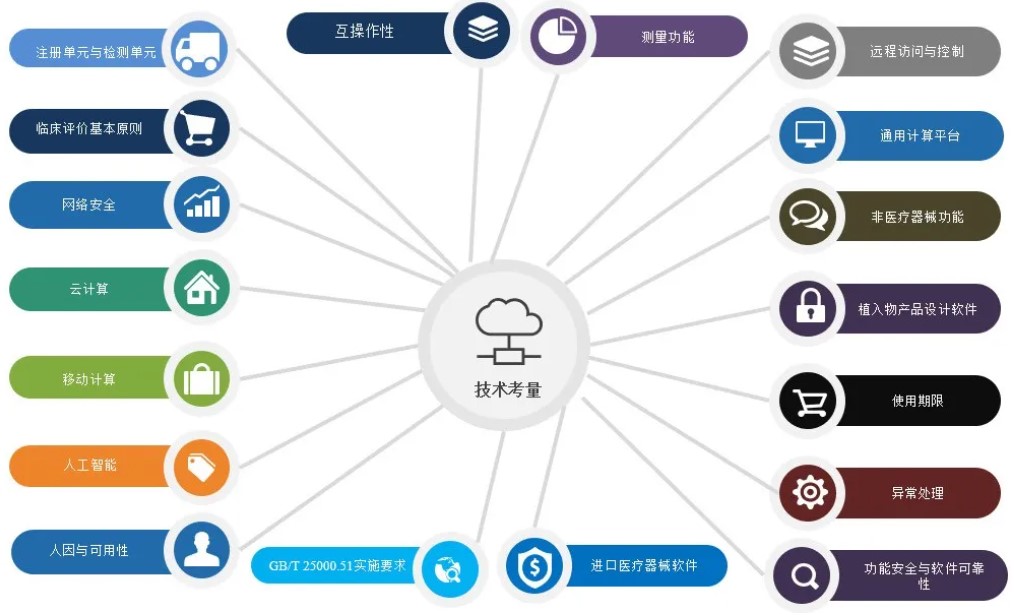
医疗质量检测技术及测试仪器展|设计技术考量点
上海医疗测试仪器展|同品种医疗器械资料是否需要获取授权?
医疗质量检测技术及测试仪器展|再谈MDR和FDA的器械可用性测试
医疗质量检测技术及测试仪器展|浅谈在用医疗器械质量检测的常见方法有哪些
医疗质量检测技术展|ISO13485和GB/T42061解读、导入与审核(条款7.3.6 设计和开发验证)
医疗质量检测技术展|FDA推荐使用的医疗器械CVSS标准
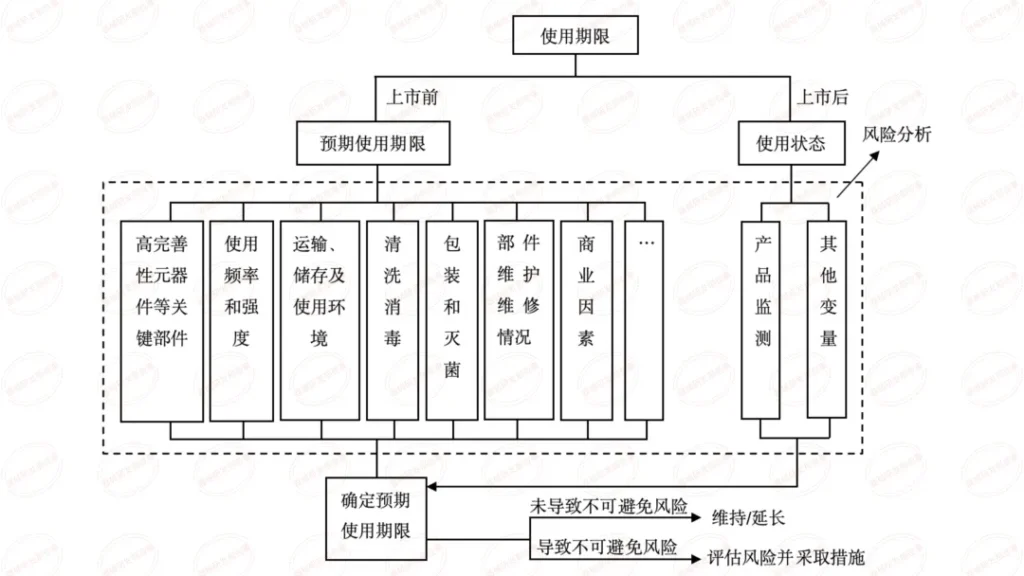
上海医疗测试仪器展|医疗器械的有效期、失效期和使用期的界定
医疗质量检测技术及测试仪器展|浅谈有源医疗器械技术审评关注点