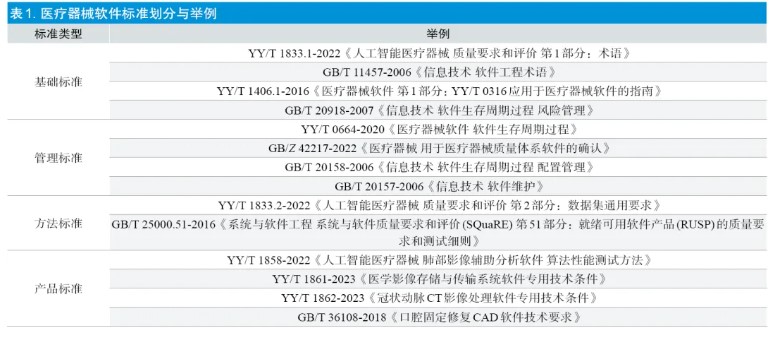
随着计算机技术的进步,医疗器械软件行业正处于高速发展期。近年来,上海市独立软件产品申报数量大幅增长。相较于其他医疗器械产品,软件产品的多样化程度高、技术发展迅速、产品迭代更新快、非实体导致缺陷隐蔽性强,因此产品的安全有效性评价难度较高。在审评核查的过程中发现,医疗器械软件产品常见基本概念不清晰、性能指标和检验方法不明确、产品验证与确认不充分、生存周期过程控制不足等问题。标准在规范产品性能指标、保证产品安全有效性、促进创新和技术进步等方面发挥着重要的作用。随着医疗器械软件行业的迅速发展以及产品自身的复杂性,对配套标准也提出了更高的要求。本文在概要论述医疗器械软件标准发展现状的基础上,分析了目前在医疗器械独立软件审评核查中与标准相关的常见问题,探讨了如何更加有效地开展医疗器械软件质量评价工作,并提出了软件标准未来发展的若干思考和建议,以期加强标准的指导意义、统一评价尺度、促进医疗器械软件的标准化进程。