



医疗质量检测技术展|FDA推荐使用的医疗器械CVSS标准

CVSS是一种普遍适用的风险评分框架,广泛用于IT和信息安全领域,帮助企业评估并优先处理各类漏洞
医疗质量检测技术及测试仪器展|US FDA关于无菌器械灭菌的要求

无菌类器械510(k)中,成熟的灭菌方法包括蒸汽灭菌、干热灭菌、环氧乙烷(EO)灭菌、辐射灭菌、汽化过氧化氢、臭氧等。正在开发的全新灭菌技术并将用于I类和II类器械生产的,被视为新方法。对于采用新灭菌技术灭菌的器械,如果操作不当,会带来灭菌不到位的重大风险。因此,应密切评估使用这些技术灭菌的器械是否符合GMP。FDA计划先审查其制造机构再授予510(k)许可,这将有助于确保器械的安全性和有效性,并降低对人类健康的风险。
医疗质量检测技术展|三类降级为二类,FDA为什么那么大的决心?
FDA对Class III检测的要求是每个经过上市前批准程序的器械都有责任独立提供数据和信息,以合理保证安全性和有效性,光是General Control和Special Control都还不够。整个过程十分漫长,涉及大量的临床试验和数据收集。FDA只有在确定足够的科学证据支持该设备对其预期用途安全有效后才会批准申请。
医疗质量检测技术及测试仪器展|再谈MDR和FDA的器械可用性测试
测试完成后,您将需要制作一份可用性工程报告,该报告需要提交给您的医疗设备销售市场的相应监管机构。需要注意的是,这份报告不应该简单地总结你的可用性验证测试,而是需要一个非常具体的格式。
上海医疗测试仪器展|医疗器械测试项目清单:确保安全合规的关键步骤

在医疗器械行业,产品的安全性和合规性一直是至关重要的问题。在上海医疗测试仪器展上,这一点尤其受到关注。为了确保医疗器械在临床使用中能够安全可靠地发挥作用,医疗器械测试是必不可少的步骤。
医疗质量检测技术展|ECA:关于欧盟无菌附录1的问答

是否有一个正式流程来确定附录1需要应用到API制造的范围?它在API制造期间如何被应用,以及如何系统地做出这些决定?
上海医疗检测设备展|医疗器械检测流程详解:哪些项目不可忽略?

医疗器械检测是确保产品安全性和有效性的关键步骤。无论是在开发阶段还是在产品上市前,医疗器械检测都扮演着至关重要的角色。企业需要通过一系列严格的检测环节,才能确保产品满足各项法规要求,顺利通过监管机构的审批,进入市场。
上海医疗检测设备展|洁净区的压差控制
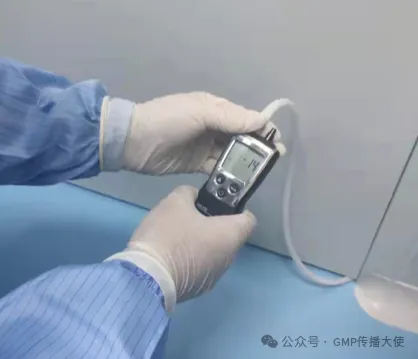
洁净室与周围的空间必须维持一定的压差,并应按工艺要求决定维持正压差,通常情况洁净度高的区域流向洁净度低的区域,使洁净室的洁净度不受到污染空气的干扰。
医疗质量检测技术展|包装运输试验和环境试验

国家药监局发布的《医疗器械安全和性能基本原则》中明确规定:医疗器械的设计、生产和包装,包括申请人所提供的说明和信息,应确保在按照预期用途使用时,运输和贮存条件(例如:跌落、振动、温度和湿度的波动)不会对医疗器械的特性和性能,包括完整性和清洁度,造成不利影响。
医疗质量检测技术及测试仪器展|医疗器械GMP要升级

2014年12月,原国家食品药品监督管理总局(CFDA)发布了医疗器械生产质量管理规范(以下简称医疗器械GMP)(2014年第64号),并随后陆续推出了无菌医疗器械、植入性医疗器械、体外诊断试剂、定制式义齿和独立软件(注:NMPA发布)等5个附录。