



医疗质量检测技术展|收藏!质量体系增加9001后,质量手册调整内容

医疗器械生产监管要求企业建立适应的质量体系以确保产品生产质量可控,但很多企业建立时仅纳入了GMP和13485,后续在做三方认证时意识到了9001的需求,故在原有体系上进行增加9001,这里就质量手册的改动部分做分享
医疗质量检测技术及测试仪器展|QMSR质量体系到底是怎么回事?

QMSR的征求意见稿于2022年的2月发布,正式版本于2024年2月发布,2026年2月2日生效,制造商有两年的时间来调整内部流程和程序。美国FDA医疗器械质量体系法规俗称为QSR820。其中820是指的是美国联邦法规的第820章节。Q代表quality,S代表system,R代表regulation。
医疗质量检测技术展|QMSR质量体系到底是怎么回事?
MSR的征求意见稿于2022年的2月发布,正式版本于2024年2月发布,2026年2月2日生效,制造商有两年的时间来调整内部流程和程序。美国FDA医疗器械质量体系法规俗称为QSR820。其中820是指的是美国联邦法规的第820章节。Q代表quality,S代表system,R代表regulation。美国FDA对QSR820质量体系进行修订,引进了国际标准化组织ISO13485-2016版本取代现有的大部分法规,更改后的质量体系新规叫做:QMSR(Quality Management System Regulation)
上海医疗测试仪器展|欧盟与我国GMP无菌药品附录差异分析

无菌药品作为高风险产品,其在生产管理和质量管理上具有其高标准和特殊性。
医疗质量检测技术展|ISO 13485条款解读、导入与审核(条款5.5.2,管理者代表)

对本条款的审核应围绕条款规定的要求进行,条款要求最高管理者指定一名管理层的成员为管代,“指定”需要具备客观的证据,因此要检查的是否有形成文件的管理者代表任命书。
医疗质量检测技术及测试仪器展|医疗器械独立软件核查中对相关标准的思考
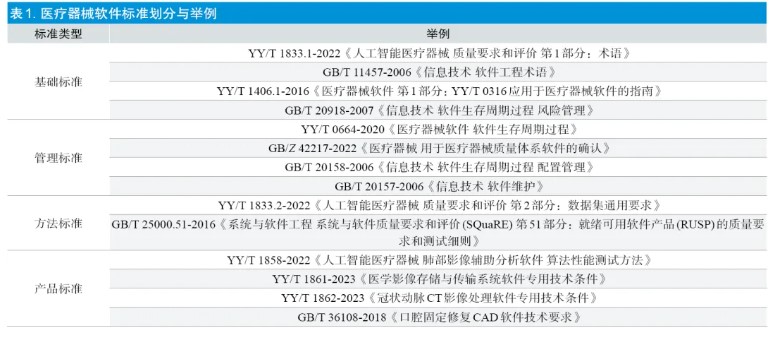
随着计算机技术的进步,医疗器械软件行业正处于高速发展期。近年来,上海市独立软件产品申报数量大幅增长。相较于其他医疗器械产品,软件产品的多样化程度高、技术发展迅速、产品迭代更新快、非实体导致缺陷隐蔽性强,因此产品的安全有效性评价难度较高。在审评核查的过程中发现,医疗器械软件产品常见基本概念不清晰、性能指标和检验方法不明确、产品验证与确认不充分、生存周期过程控制不足等问题。标准在规范产品性能指标、保证产品安全有效性、促进创新和技术进步等方面发挥着重要的作用。随着医疗器械软件行业的迅速发展以及产品自身的复杂性,对配套标准也提出了更高的要求。本文在概要论述医疗器械软件标准发展现状的基础上,分析了目前在医疗器械独立软件审评核查中与标准相关的常见问题,探讨了如何更加有效地开展医疗器械软件质量评价工作,并提出了软件标准未来发展的若干思考和建议,以期加强标准的指导意义、统一评价尺度、促进医疗器械软件的标准化进程。
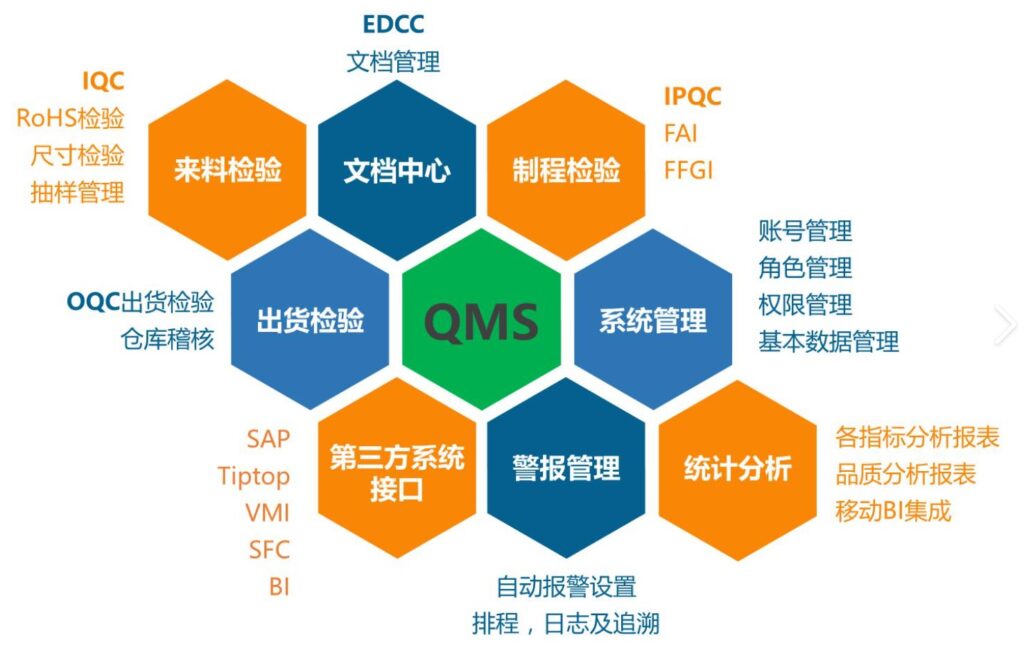
医疗质量检测技术展|医疗器械生产组织的质量管理体系需满足的四方面要求

医疗器械生产组织的质量管理体系需满足四方面要求,即质量管理体系GB/T42061标准要求、法规要求、顾客要求和组织自身要求。
上海医疗检测设备展|医疗器械认证过程中的风险管理要点

当我们谈论医疗器械行业的风险管理时,有些人可能会认为这是一个令人生畏的混乱过程。一开始可能会让人不知所措,但这对确保我们开发的器械在整个生命周期内安全有效至关重要。这不仅仅是为了满足监管要求—虽然这绝对是其中的一部分—而是为了做出明智的决策,保护患者并降低器械失效的可能性。
医疗质量检测技术展|【法规解读】欧盟公布IVDR过渡期延长条例,看普瑞专家如何解读,给出策略

IVDR过渡期延长的条例终于于2024.7.9颁布了。那么来看看过渡期延长后,制造商应做如何应对。
Quality Expo|什么医疗器械产品可以免于注册检测?

注册证是进入市场的入场券。在我国,第二、三类医疗器械要投入销售、使用,须先按照《医疗器械注册管理办法》或《体外诊断试剂注册管理办法》的相关规定向食药监部门申请产品注册,取得医疗器械注册批件,从而获得进入市场的资格。