



医疗质量检测技术展|中国GMP医疗器械生产质量管理规范包含哪些?
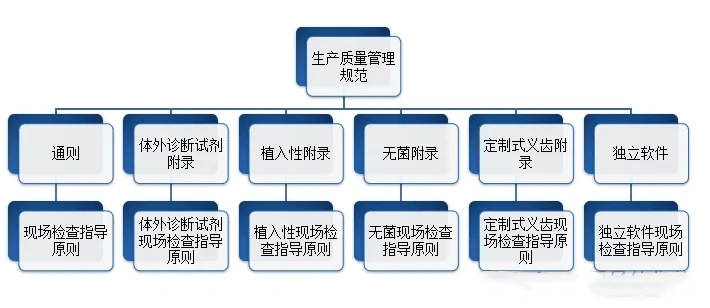
生产企业应当按照医疗器械生产质量管理规范的要求,结合产品特点,建立健全与所生产医疗器械相适应的质量管理体系,并保证其有效运行。在医疗质量检测技术展上,这些生产企业将有机会展示他们如何遵循这些规范,并通过先进的质量管理系统确保其产品的高标准和可靠性。
上海医疗测试仪器展|2024年最新有源医疗器械风险管理的流程
有源医疗器械在医疗领域中扮演着至关重要的角色,而风险管理则是确保其安全、有效和质量可控的关键环节。以下将详细介绍有源医疗器械风险管理的流程。
医疗质量检测技术展|医疗器械必学法规:医疗器械使用质量监督管理办法(国家食药监管理总局令第18号)

这份法规文件主要规定了医疗器械在使用环节的质量管理要求和监督管理措施,旨在确保医疗器械的使用安全、有效。以下是文件中的关键知识点:
医疗质量检测技术展|ISO 13485条款解读、导入与审核(条款5.5.2,管理者代表)

对本条款的审核应围绕条款规定的要求进行,条款要求最高管理者指定一名管理层的成员为管代,“指定”需要具备客观的证据,因此要检查的是否有形成文件的管理者代表任命书。
医疗质量检测技术及测试仪器展|质量风险管理Quality Risk Management⟴8. Events and Consequences 事件与后果

本章探讨风险管理中至关重要的事件与后果的概念。理解这些要素对于准确评估和减轻风险至关重要。我们将探讨ISO 31000:2018提供的定义,审视常见的误解,并强调正确识别和分析潜在结果的重要性。通过掌握这些概念,风险专业人士可以确保更可靠和有效的分析。
医疗质量检测技术及测试仪器展|洁净室验证管理规范

本规范基于《医药工业洁净厂房设计规范》,进一步细化了医疗器械生产洁净室的验证管理要求,特别是设计计划(DQ)、安装验证(IQ)、运行验证(OQ)和性能验证(PQ)四个关键阶段,以确保洁净室的设计、建设、运行均符合GMP及国家相关标准,保障医疗器械的生产质量和安全性。
医疗质量检测技术及测试仪器展|研发用仪器、仪表计量/校准/校验的管理
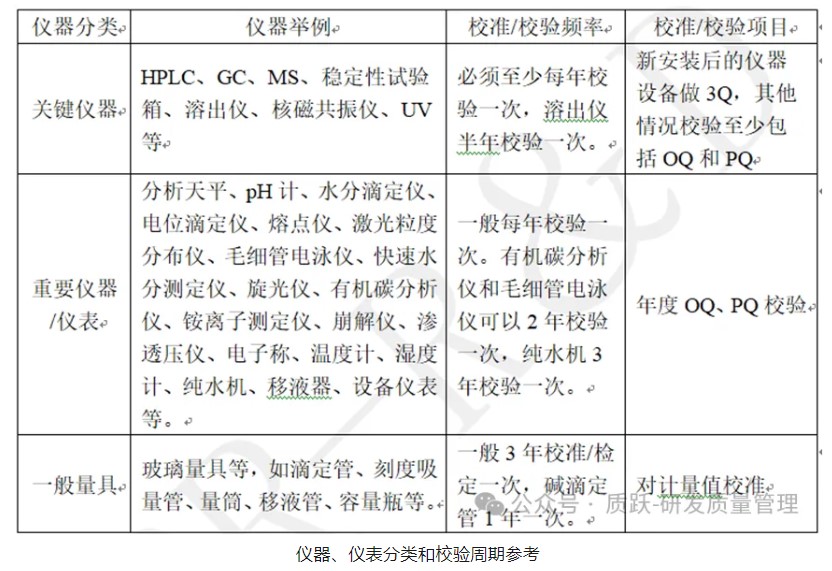
为确保研发试验数据的准确可靠,研发使用的仪器、仪表需要进行计量/校准/校验。不同的仪器、仪表计量/校准/校验的要求是有所不同的。按仪器、仪表精度和故障对数据或产品的影响程度,分为:关键仪器、重要仪器/仪表和一般量具。
医疗质量检测技术展|医疗器械生产组织的质量管理体系需满足的四方面要求

医疗器械生产组织的质量管理体系需满足四方面要求,即质量管理体系GB/T42061标准要求、法规要求、顾客要求和组织自身要求。
医疗质量检测技术展|医疗器械注册质量管理体系核查指南介绍

本指南旨在加强医疗器械注册质量管理体系的核查管理,确保核查工作的质量。依据包括《医疗器械监督管理条例》、《医疗器械注册与备案管理办法》、《体外诊断试剂注册与备案管理办法》、《医疗器械生产监督管理办法》、《医疗器械生产质量管理规范》、《医疗器械临床试验质量管理规范》及《医疗器械注册自检管理规定》等法规。
上海医疗测试仪器展|ISO13485医疗器械质量管理标准 问与答 (一)

医疗器械质量管理体系起源于2000年我国发布的第22号医疗器械质量管理体系核查要求,并在此基础上历经九年发展形成了我国第一版医疗器械生产质量管理规范。随着医疗器械行业的快速发展,我国于2014年对GMP进行了修正并形成了最新的标准规范。同时,其他国家和地区也陆续建立了相应的质量管理体系标准法规,例如美国的CDMP及后续QSF20法规,欧盟的ENISO13485标准及其多次修订等。在70年代,美国率先制定了CDMP法规,并在其基础上于1996年发布针对质量管理体系要求的QSF20法规。欧盟则从1993年开始酝酿并报告了EN1N46001标准法规,但后来对该标准进行了修订和完善。国际标准化组织在1996年发布了基于ISO9001系列的两个医疗器械质量管理体系标准,即ISO13485:1996版与ISO13481:1996版,并随9001版本升级不断调整优化,最终在2016年发布了全新的ISO13485:2016版标准。