



医疗质量检测技术及测试仪器展|医疗器械生物相容性简介、流程、路径和数据
医疗器械生物相容性评价是确保医疗器械安全性和有效性的重要环节,它涉及到对医疗器械在与人体接触时可能产生的生物学反应和风险的全面评估。根据国家标准GB/T 16886.1-2022《医疗器械生物学评价 第1部分:风险管理过程中的评价与试验》,生物相容性评价应作为医疗器械总体评价和开发过程的一部分,贯穿于产品的全生命周期。
医疗质量检测技术展|医械生物相容性评价的目标和方法

根据ISO 10993-1:2018的定义,生物相容性是指医疗器械或材料在一个特定应用中引起恰当宿主反应的能力。“生物相容”与“生物不相容”不是某种材料天然的或绝对的“标签”,而是需要结合材料的具体性能和特定的(临床)应用场景进行判断。同时,生物相容性是一个动态概念,植入物植入人体后会对特定生物组织环境产生物理和化学影响,引起生物学反应;反之,生物组织也会对植入物产生影响,使之发生物理或化学变化,两者的相互作用会一直持续。即使植入物被完全去除或被人体完全吸收,其影响还将持续一段时间。
医疗质量检测技术展|医疗器械生物相容性评价介绍

医疗器械生物相容性评价是一个复杂而细致的过程,它要求对医疗器械的各个方面进行全面考量。随着科技的发展和标准的完善,生物相容性评价将继续发展,以确保医疗器械的安全性和有效性,保护患者的健康。
医疗器械生物相容性评价

生物相容性作为保障医疗器械安全性的一个重要指标,其研究分析必须贯穿项目的始终,占据了大量财力、精力和时间。很多行业人员谈“生物相容性”即色变,其实懂得了原理,生物相容性评价也很简单。医疗器械立项、研发等早期的过程中,生物相容性所需要的成本,基本上可以转移至上游供应商,要求他们提供相应的证明资料,并签订质量协议规定供方的责任和风险。
医疗质量检测技术及测试仪器展|浅谈有源医疗器械技术审评关注点
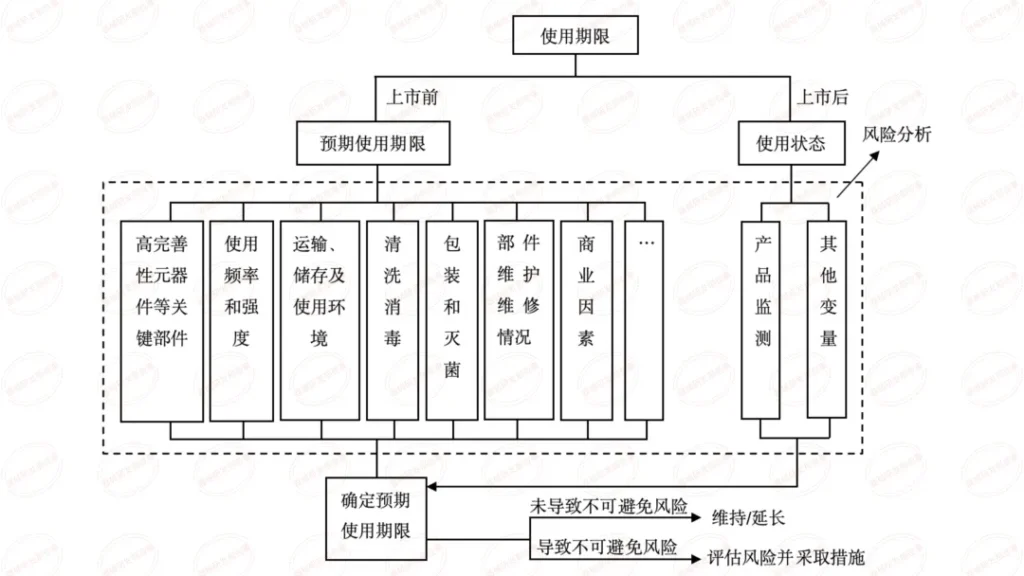
本篇内容为学习总结,源自审评二部陈敏老师关于《有源医疗器械注册申报资料要求及常见问题介绍》在器审中心学习云课程的视频。陈老师主要介绍了共性问题、研究资料-联合使用、研究资料-生物学特性、研究资料-清洁、消毒、灭菌、稳定性和92个技术审评共性问题等六个方面。
医疗质量检测技术及测试仪器展|验证与确认:医疗器械领域的深入解析与实例
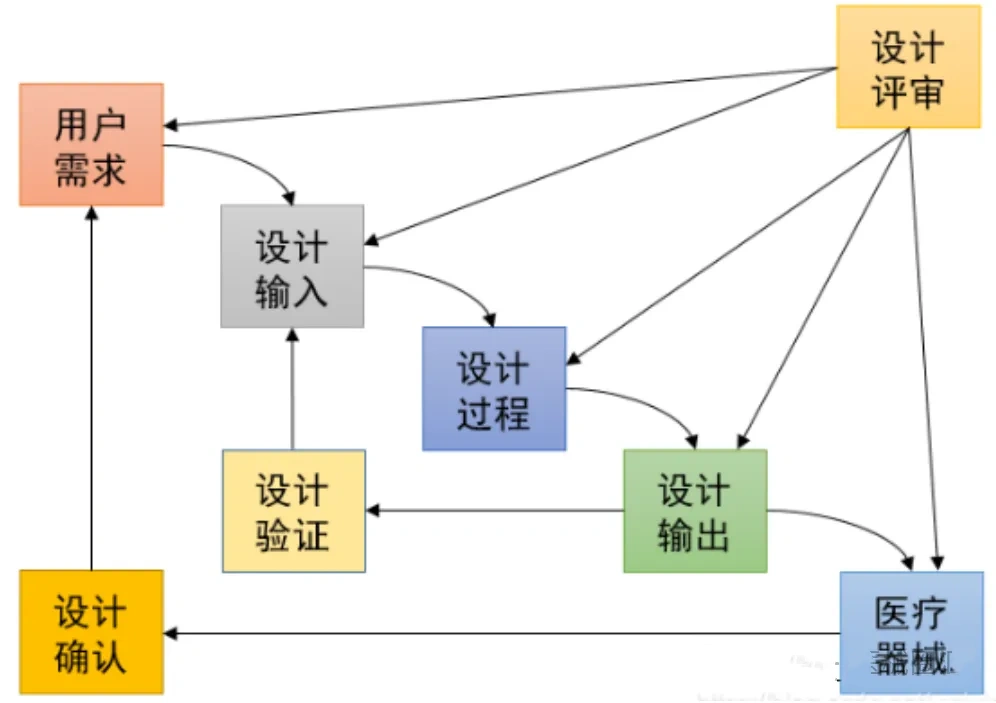
在医疗器械行业,验证(Verification)与确认(Validation)不仅仅是质量管理的基本要素,更是确保产品安全、有效性的关键步骤。在医疗质量检测技术及测试仪器展上,这些关键步骤得到了特别的关注和展示。
上海医疗测试仪器展|医疗器械测试项目清单:确保安全合规的关键步骤

在医疗器械行业,产品的安全性和合规性一直是至关重要的问题。在上海医疗测试仪器展上,这一点尤其受到关注。为了确保医疗器械在临床使用中能够安全可靠地发挥作用,医疗器械测试是必不可少的步骤。
医疗质量检测技术展|浅谈在用医疗器械质量检测的常见方法有哪些

在用医疗器械质量检测的常见方法多种多样,这些方法旨在确保医疗器械的安全性、有效性和符合相关标准。
上海医疗检测设备展|医疗器械检测流程详解:哪些项目不可忽略?

医疗器械检测是确保产品安全性和有效性的关键步骤。无论是在开发阶段还是在产品上市前,医疗器械检测都扮演着至关重要的角色。企业需要通过一系列严格的检测环节,才能确保产品满足各项法规要求,顺利通过监管机构的审批,进入市场。
上海医疗检测设备展|无菌医疗器械包装质量控制要点(下)

无菌包装生产技术已经成熟。随着法规和检测技术的不断完善,使用企业对质量要求在逐步提高。法规和质量标准的推出将进一步规范无菌包装产品的质量和性能,从而更好地保障无菌医疗器械的安全性和有效性。