



上海医疗测试仪器展|医疗器械生物学评价流程与思路
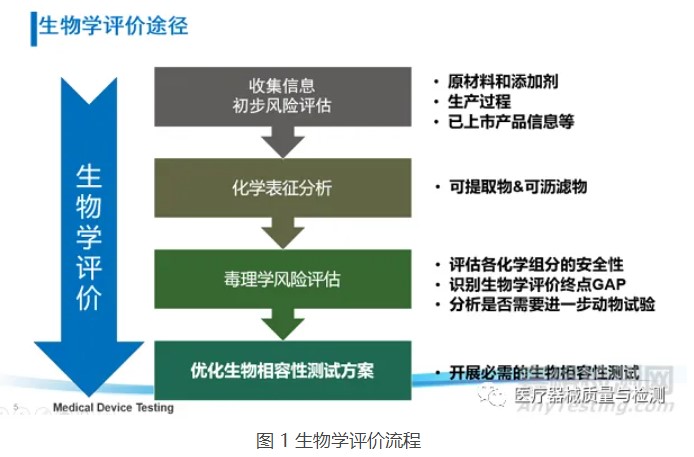
考虑器械潜在的生物学风险,并不意味着要针对所有风险点都开展生物学测试,还可通过“评价”的方式开展物理/化学表征及毒理学风险评估、基于已有的临床应用历史和人体接触数据对器械的生物学风险进行评估。在近年来更新的ISO 10993-1:2018和FDA年发布的关于ISO 10993-1的应用指南中,均强调了通过化学表征测试和毒理学风险评估(ISO 10993-18和ISO 10993-17)进行生物学评价的思路,不仅可以豁免不必要的生物相容性测试及避免人力、物力和动物资源的浪费,还可以基于已有的研究数据更加充分地评估器械(尤其是持久性植入的高风险类器械)中潜在的生物安全性风险(如慢性毒性、致癌性和生殖毒性等),从而优化生物学测试方案,最终达到器械安全性评价的目的。
医疗质量检测技术及测试仪器展|浅谈有源医疗器械技术审评关注点
本篇内容为学习总结,源自审评二部陈敏老师关于《有源医疗器械注册申报资料要求及常见问题介绍》在器审中心学习云课程的视频。陈老师主要介绍了共性问题、研究资料-联合使用、研究资料-生物学特性、研究资料-清洁、消毒、灭菌、稳定性和92个技术审评共性问题等六个方面。
上海医疗检测设备展|无菌医疗器械包装质量控制要点(下)

无菌包装生产技术已经成熟。随着法规和检测技术的不断完善,使用企业对质量要求在逐步提高。法规和质量标准的推出将进一步规范无菌包装产品的质量和性能,从而更好地保障无菌医疗器械的安全性和有效性。
医疗质量检测技术及测试仪器展|无菌医疗器械包装质量控制要点(上)
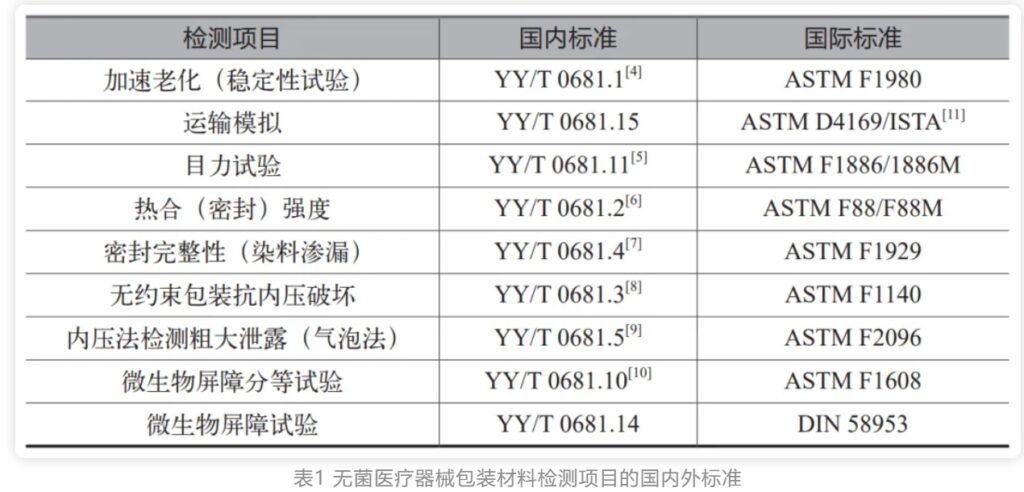
无菌医疗器械包装是保护医疗器械、预防感染的最后一道防线,包装失效会对病人和护士人员的健康和生命带来威胁。无菌医疗器械包装也称为无菌包装屏障系统,不同于食品行业的无菌包装。医疗器械无菌包装不仅需要有阻隔微生物的屏障能力,还要确保灭菌后能在一定期限内维持器械的无菌状态,并能够经受如环氧乙烷、伽马辐照、等离子过氧化氢、蒸汽等方式的灭菌。因此无菌医疗器械包装也被认为是医疗器械组成的一部分。
医疗质量检测技术展|医疗器械原材料变更的基本流程

医疗器械的预期用途、技术要求、安全有效性、有效期等等,很大程度上取决于原材料。
产品注册时确定下来的原材料,轻易不能变化。
但是,企业在实际经营过程中,常常为了降低成本、改善某方面性能、完善供应链、符合升版的标准,或主动、或被动地对原材料进行变更。
医疗质量检测技术及测试仪器展|医疗器械生物相容性简介、流程、路径和数据
医疗器械生物相容性评价是确保医疗器械安全性和有效性的重要环节,它涉及到对医疗器械在与人体接触时可能产生的生物学反应和风险的全面评估。根据国家标准GB/T 16886.1-2022《医疗器械生物学评价 第1部分:风险管理过程中的评价与试验》,生物相容性评价应作为医疗器械总体评价和开发过程的一部分,贯穿于产品的全生命周期。
医疗质量检测技术展|医疗器械注册检验常见问题答疑汇总

A:维氏硬度检测不需要提供单独的试样,在送检样品上取样测试;洛氏硬度检测若被测部位形状不规则时,无法直接对样品进行测试,应提供原材料试样块,并提供材质、热处理工艺一致性声明。
医疗质量检测技术展|医械生物相容性评价的目标和方法

根据ISO 10993-1:2018的定义,生物相容性是指医疗器械或材料在一个特定应用中引起恰当宿主反应的能力。“生物相容”与“生物不相容”不是某种材料天然的或绝对的“标签”,而是需要结合材料的具体性能和特定的(临床)应用场景进行判断。同时,生物相容性是一个动态概念,植入物植入人体后会对特定生物组织环境产生物理和化学影响,引起生物学反应;反之,生物组织也会对植入物产生影响,使之发生物理或化学变化,两者的相互作用会一直持续。即使植入物被完全去除或被人体完全吸收,其影响还将持续一段时间。
医疗质量检测技术及测试仪器展 | 浅谈吻(缝)合器外科产品技术评审要点
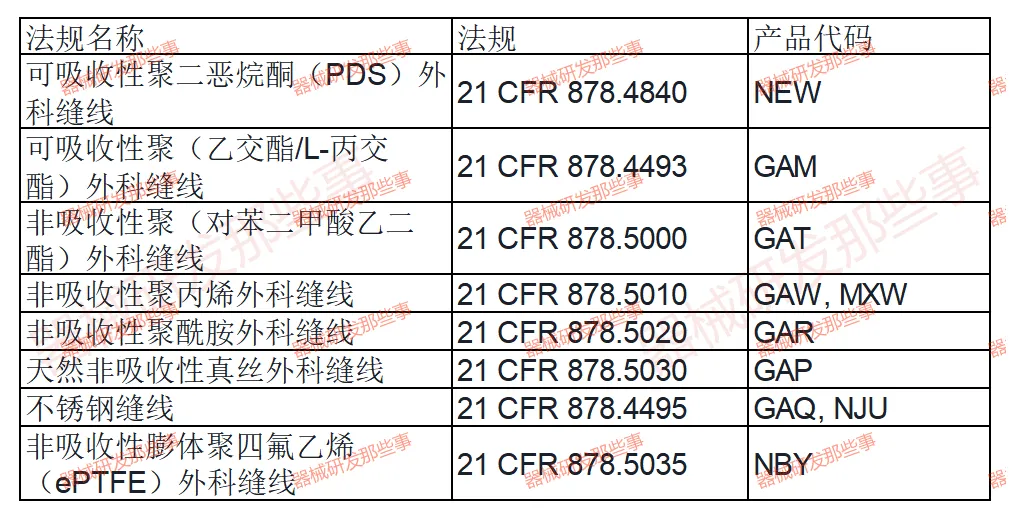
吻(缝)合器外科产品在临床手术中扮演着至关重要的角色,其安全性、有效性和可靠性直接关系到患者的生命健康。
医疗质量检测技术及测试仪器展 | 一大波国内外领先的质量检测供应商来袭!来Medtec 2024现场一探究竟(一)
医疗质量检测技术及测试仪器展整理了部分质量检测展品预览,快来一睹为快吧~