



上海医疗测试仪器展|2024年最新有源医疗器械风险管理的流程
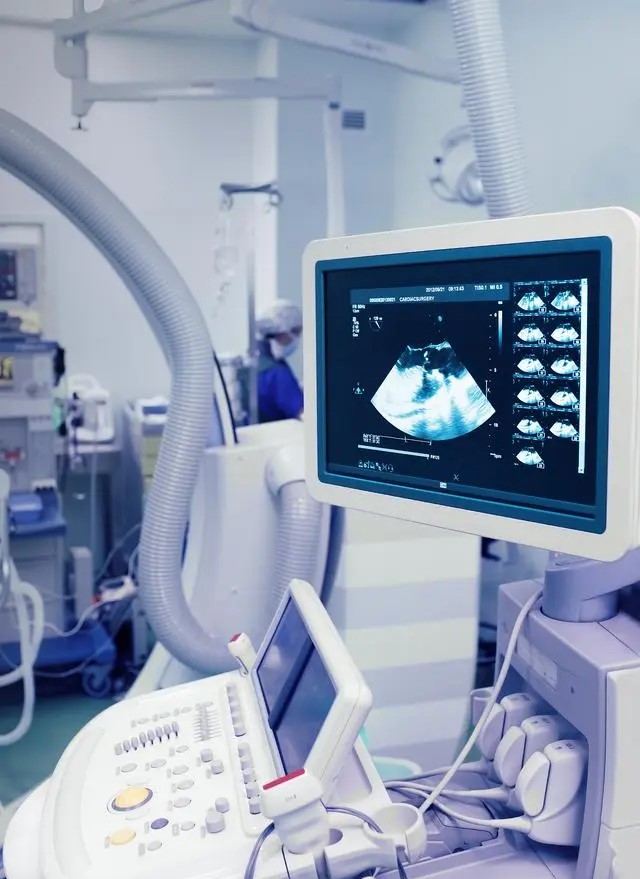
有源医疗器械在医疗领域中扮演着至关重要的角色,而风险管理则是确保其安全、有效和质量可控的关键环节。以下将详细介绍有源医疗器械风险管理的流程。
上海医疗检测设备展|一文了解医疗器械检测的重要性和流程

医疗器械检测在保障患者安全、提升医疗质量以及推动医疗器械行业健康发展方面扮演着至关重要的角色。上海医疗检测设备 […]
上海医疗检测设备展|医疗器械检测流程详解:哪些项目不可忽略?

医疗器械检测是确保产品安全性和有效性的关键步骤。无论是在开发阶段还是在产品上市前,医疗器械检测都扮演着至关重要的角色。企业需要通过一系列严格的检测环节,才能确保产品满足各项法规要求,顺利通过监管机构的审批,进入市场。
医疗质量检测技术展|医疗器械原材料变更的基本流程

医疗器械的预期用途、技术要求、安全有效性、有效期等等,很大程度上取决于原材料。
产品注册时确定下来的原材料,轻易不能变化。
但是,企业在实际经营过程中,常常为了降低成本、改善某方面性能、完善供应链、符合升版的标准,或主动、或被动地对原材料进行变更。
医疗质量检测技术及测试仪器展|医疗器械生物相容性简介、流程、路径和数据
医疗器械生物相容性评价是确保医疗器械安全性和有效性的重要环节,它涉及到对医疗器械在与人体接触时可能产生的生物学反应和风险的全面评估。根据国家标准GB/T 16886.1-2022《医疗器械生物学评价 第1部分:风险管理过程中的评价与试验》,生物相容性评价应作为医疗器械总体评价和开发过程的一部分,贯穿于产品的全生命周期。
上海医疗测试仪器展|医疗器械生物学评价流程与思路
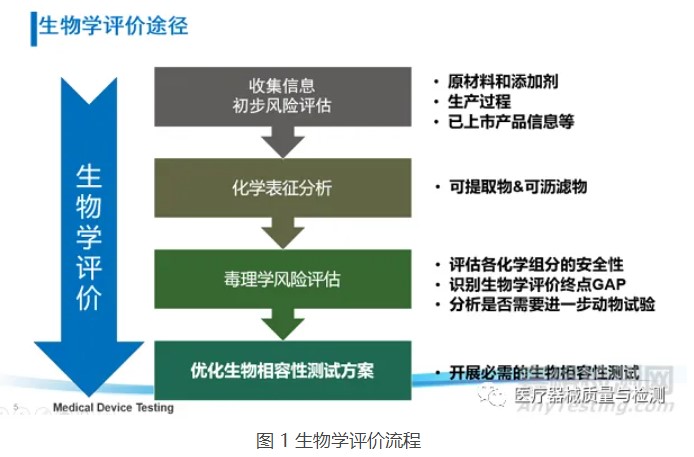
考虑器械潜在的生物学风险,并不意味着要针对所有风险点都开展生物学测试,还可通过“评价”的方式开展物理/化学表征及毒理学风险评估、基于已有的临床应用历史和人体接触数据对器械的生物学风险进行评估。在近年来更新的ISO 10993-1:2018和FDA年发布的关于ISO 10993-1的应用指南中,均强调了通过化学表征测试和毒理学风险评估(ISO 10993-18和ISO 10993-17)进行生物学评价的思路,不仅可以豁免不必要的生物相容性测试及避免人力、物力和动物资源的浪费,还可以基于已有的研究数据更加充分地评估器械(尤其是持久性植入的高风险类器械)中潜在的生物安全性风险(如慢性毒性、致癌性和生殖毒性等),从而优化生物学测试方案,最终达到器械安全性评价的目的。
医疗质量检测技术展|FDA推荐使用的医疗器械CVSS标准

CVSS是一种普遍适用的风险评分框架,广泛用于IT和信息安全领域,帮助企业评估并优先处理各类漏洞
上海医疗检测设备展|医疗器械工艺用水验证

通过对工艺用水系统的全面验证,确保该系统能够连续稳定地供应满足生产需求的水量,并证明制水过程能够在长期内持续制备出符合《医疗器械生产质量管理规范-无菌医疗器械实施细则》要求的工艺用水。
医疗质量检测技术及测试仪器展|验证与确认:医疗器械领域的深入解析与实例
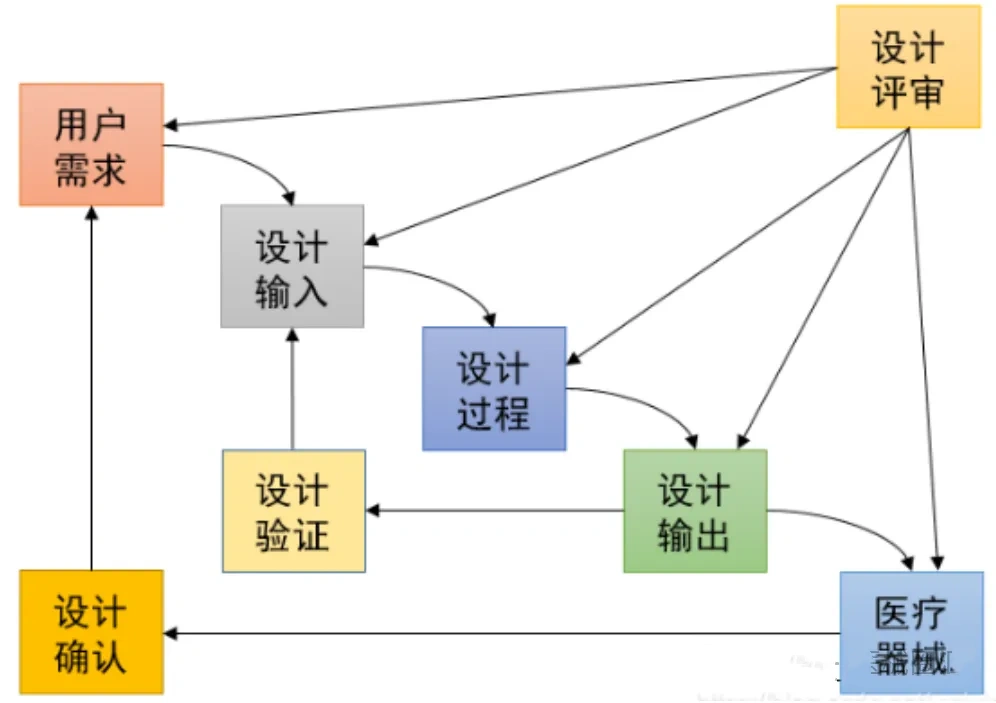
在医疗器械行业,验证(Verification)与确认(Validation)不仅仅是质量管理的基本要素,更是确保产品安全、有效性的关键步骤。在医疗质量检测技术及测试仪器展上,这些关键步骤得到了特别的关注和展示。
上海医疗检测设备展|ISO13485和GB/T42061解读、导入与审核(条款7.3.3 设计和开发输入)
ISO13485:2016《医疗器械质量管理体系用于法规的要求》,由ISO国际标准化组织于2016年3月1 日正式发布,是一项应用于医疗器械领域的质量管理体系的国际标准,强调医疗器械的安全有效,组织提供的医疗器械要满足顾客和法规要求。