



医疗质量检测技术及测试仪器展|无源医疗器械安全性评价第二部分:货架有效期
医疗器械注册阶段,货架有效期部分研究至关重要,货架有效期研究是确保产品在规定期限内能够正常发挥预期功能的重要证据。当医疗器械超过货架有效期研究规定的最大期限,器械的性能可能无法得到保证,在使用时则存在潜在风险。
上海医疗测试仪器展|医疗器械安全性评价的总体框架要点梳理

医疗器械安全性评价的内容较为繁杂,安全性评价有共性部分,也有差异部分,产品不同,评价内容不同,评价重点不同。
识别安全性问题的来源,是进行安全性评价的基础。医界内参老师整理了安全性评价的整体思路和框架,加入了我平台老师自己的经验和理解,以促进从业朋友对医疗器械安全性评价内容的概要理解。
医疗质量检测技术及测试仪器展|洁净室验证管理规范

本规范基于《医药工业洁净厂房设计规范》,进一步细化了医疗器械生产洁净室的验证管理要求,特别是设计计划(DQ)、安装验证(IQ)、运行验证(OQ)和性能验证(PQ)四个关键阶段,以确保洁净室的设计、建设、运行均符合GMP及国家相关标准,保障医疗器械的生产质量和安全性。
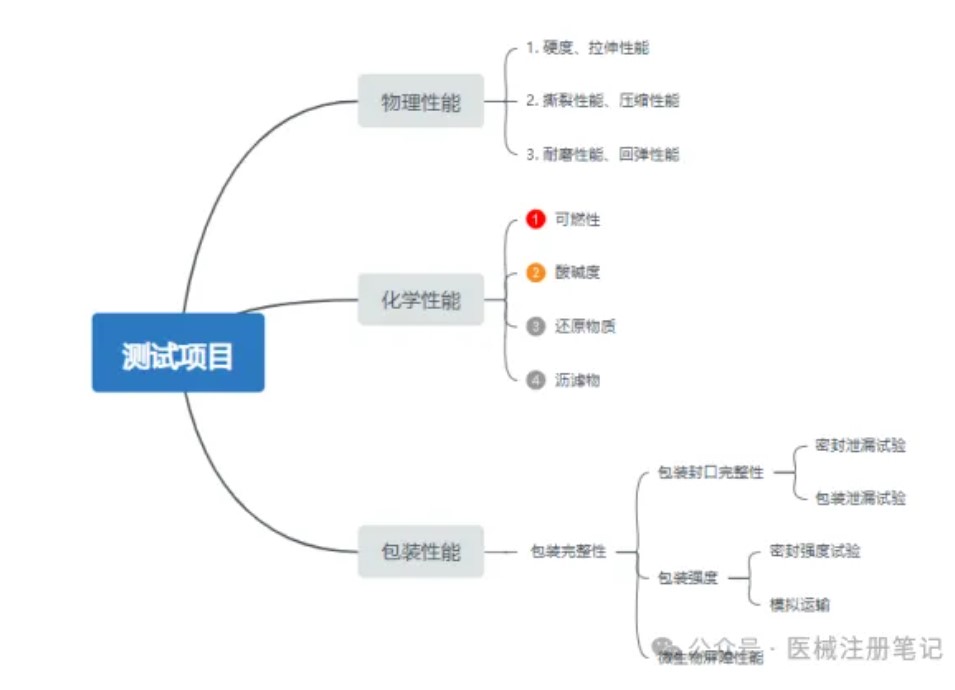
医疗质量检测技术及测试仪器展|无菌医疗器械包装质量控制要点(上)
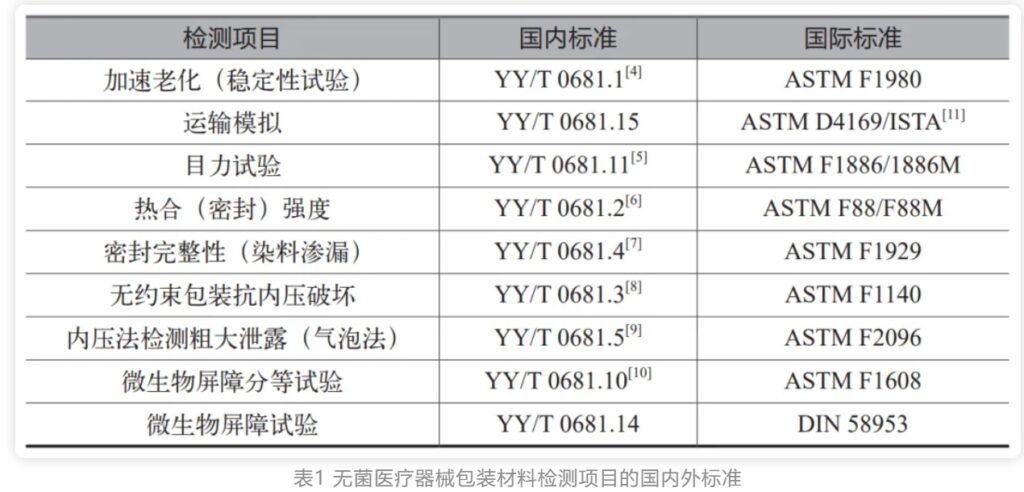
无菌医疗器械包装是保护医疗器械、预防感染的最后一道防线,包装失效会对病人和护士人员的健康和生命带来威胁。无菌医疗器械包装也称为无菌包装屏障系统,不同于食品行业的无菌包装。医疗器械无菌包装不仅需要有阻隔微生物的屏障能力,还要确保灭菌后能在一定期限内维持器械的无菌状态,并能够经受如环氧乙烷、伽马辐照、等离子过氧化氢、蒸汽等方式的灭菌。因此无菌医疗器械包装也被认为是医疗器械组成的一部分。
医疗质量检测技术展|医疗器械软件注册检验要点及方法研究
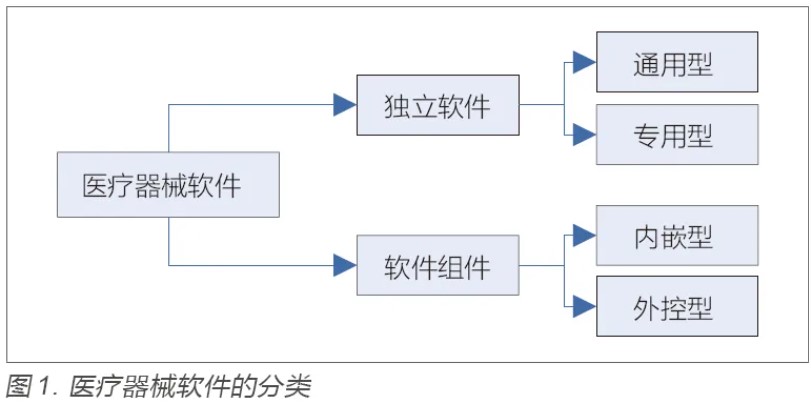
目的:针对医疗器械软件注册检验要点和检测方法进行研究,为加强医疗器械软件的质量保证工作提供参考。方法:以医疗器械软件注册指导原则为基础,梳理不同医疗器械软件的特点和分类,明确软件产品技术要求的编制指南,并且结合通用要求、质量要求、专用要求、安全要求四个方面的要求,对注册检验内容进行解读,最后总结常用检测方法和检测工具。结果:梳理的医疗器械软件注册检验要点可以为注册申请人编写产品技术要求提供指导,总结的检验方法和检测工具可以给检测机构开展相关评测提供借鉴。结论:医疗器械软件检验应从通用要求、质量要求、专用要求和安全要求四个方面进行,检测主要涉及功能测试和性能测试两大类,性能测试通常需借助专业工具进行。
医疗质量检测技术及测试仪器展|US FDA关于无菌器械灭菌的要求

无菌类器械510(k)中,成熟的灭菌方法包括蒸汽灭菌、干热灭菌、环氧乙烷(EO)灭菌、辐射灭菌、汽化过氧化氢、臭氧等。正在开发的全新灭菌技术并将用于I类和II类器械生产的,被视为新方法。对于采用新灭菌技术灭菌的器械,如果操作不当,会带来灭菌不到位的重大风险。因此,应密切评估使用这些技术灭菌的器械是否符合GMP。FDA计划先审查其制造机构再授予510(k)许可,这将有助于确保器械的安全性和有效性,并降低对人类健康的风险。
医疗质量检测技术展|三类降级为二类,FDA为什么那么大的决心?
FDA对Class III检测的要求是每个经过上市前批准程序的器械都有责任独立提供数据和信息,以合理保证安全性和有效性,光是General Control和Special Control都还不够。整个过程十分漫长,涉及大量的临床试验和数据收集。FDA只有在确定足够的科学证据支持该设备对其预期用途安全有效后才会批准申请。
医疗质量检测技术展|人工智能医疗器械注册审查基本原则
人工智能算法的类型不同,其算法特性、适用场景也不同,评价重点亦有所侧重;同时,不同类型的人工智能算法可组合使用,需结合各算法特性和算法组合形式进行整体评价。
医疗质量检测技术及测试仪器展|网络安全更新
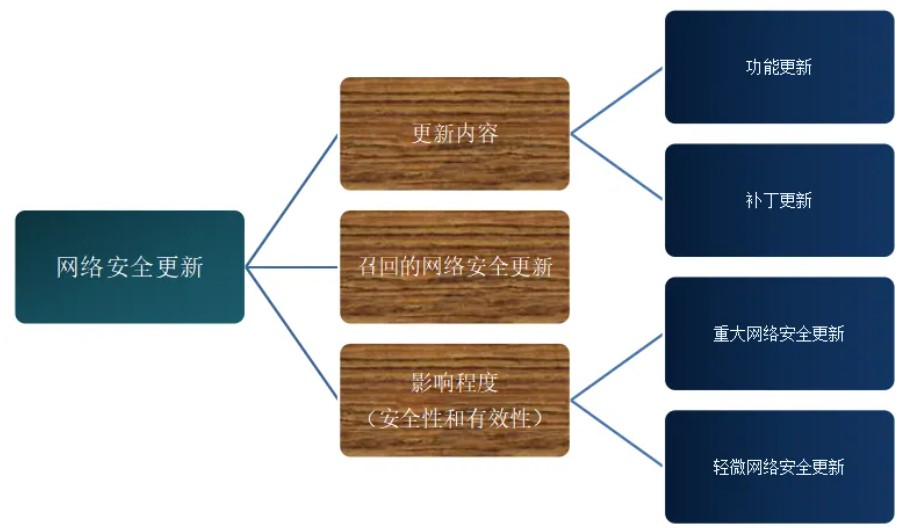
医疗器械网络安全更新从内容上可分为功能更新、补丁更新,类似于增强类软件更新、纠正类软件更新。
根据其对医疗器械安全性和有效性的影响程度分为以下两类:
医疗质量检测技术及测试仪器展|设计技术考量点
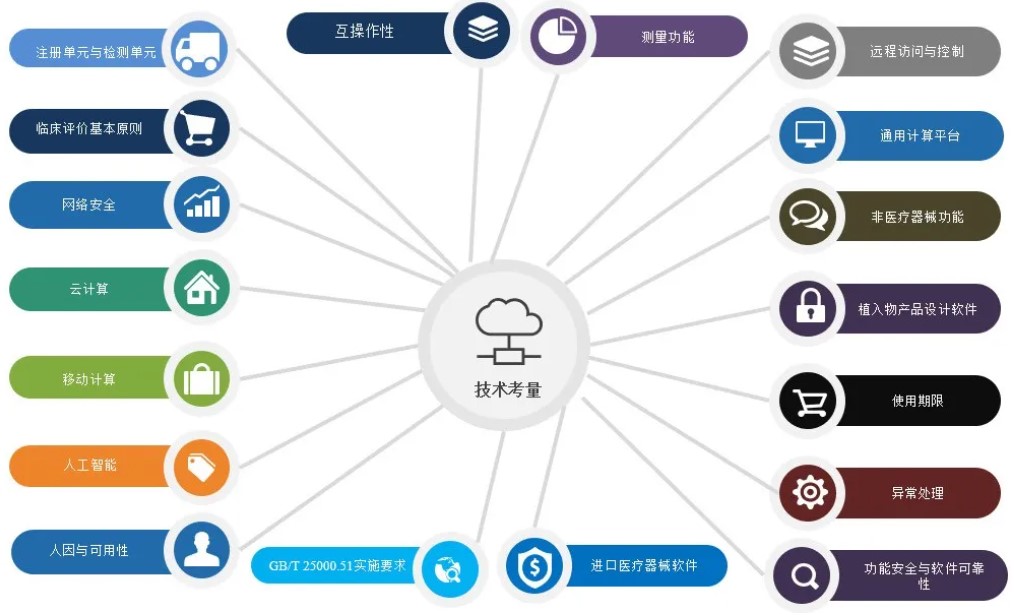
在进行医疗期器械软件设计过程中,我们可以从以下18点来进行技术考量。分别为1.注册单元与检测单元、2.临床评价基本原则、3.网络安全、4.云计算、5.移动计算、6.人工智能、7.人因与可用性、8.互操作性、9.测量功能、10.远程访问与控制、11.通用计算平台、12.非医疗器械功能、13.植入产品设计软件、14.使用期限、15.异常处理、16.功能安全与软件可靠性、17.GB/T25000.51实施要求和18.进口医疗器械软件等要求。