



医疗质量检测技术展|医疗器械注册质量管理体系核查指南介绍

本指南旨在加强医疗器械注册质量管理体系的核查管理,确保核查工作的质量。依据包括《医疗器械监督管理条例》、《医疗器械注册与备案管理办法》、《体外诊断试剂注册与备案管理办法》、《医疗器械生产监督管理办法》、《医疗器械生产质量管理规范》、《医疗器械临床试验质量管理规范》及《医疗器械注册自检管理规定》等法规。
医疗质量检测技术及测试仪器展|医疗器械独立软件核查中对相关标准的思考
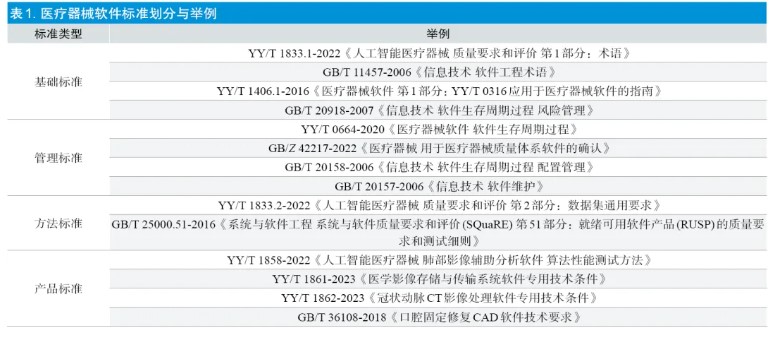
随着计算机技术的进步,医疗器械软件行业正处于高速发展期。近年来,上海市独立软件产品申报数量大幅增长。相较于其他医疗器械产品,软件产品的多样化程度高、技术发展迅速、产品迭代更新快、非实体导致缺陷隐蔽性强,因此产品的安全有效性评价难度较高。在审评核查的过程中发现,医疗器械软件产品常见基本概念不清晰、性能指标和检验方法不明确、产品验证与确认不充分、生存周期过程控制不足等问题。标准在规范产品性能指标、保证产品安全有效性、促进创新和技术进步等方面发挥着重要的作用。随着医疗器械软件行业的迅速发展以及产品自身的复杂性,对配套标准也提出了更高的要求。本文在概要论述医疗器械软件标准发展现状的基础上,分析了目前在医疗器械独立软件审评核查中与标准相关的常见问题,探讨了如何更加有效地开展医疗器械软件质量评价工作,并提出了软件标准未来发展的若干思考和建议,以期加强标准的指导意义、统一评价尺度、促进医疗器械软件的标准化进程。
医疗质量检测技术展|人工智能医疗器械注册审查基本原则
人工智能算法的类型不同,其算法特性、适用场景也不同,评价重点亦有所侧重;同时,不同类型的人工智能算法可组合使用,需结合各算法特性和算法组合形式进行整体评价。
医疗质量检测技术展|医疗器械“体考”到底是什么?

《医疗器械生产监督管理办法》明确规定:医疗器械生产企业应当按照医疗器械生产质量管理规范的要求,建立质量管理体系并保持有效运行。申请人在申请产品注册时,受理注册申请的药品监督管理部门在产品技术审评时,认为有必要对质量管理体系进行核查的,会组织开展质量管理体系核查。
上海医疗测试仪器展|ISO13485医疗器械质量管理标准 问与答 (一)

医疗器械质量管理体系起源于2000年我国发布的第22号医疗器械质量管理体系核查要求,并在此基础上历经九年发展形成了我国第一版医疗器械生产质量管理规范。随着医疗器械行业的快速发展,我国于2014年对GMP进行了修正并形成了最新的标准规范。同时,其他国家和地区也陆续建立了相应的质量管理体系标准法规,例如美国的CDMP及后续QSF20法规,欧盟的ENISO13485标准及其多次修订等。在70年代,美国率先制定了CDMP法规,并在其基础上于1996年发布针对质量管理体系要求的QSF20法规。欧盟则从1993年开始酝酿并报告了EN1N46001标准法规,但后来对该标准进行了修订和完善。国际标准化组织在1996年发布了基于ISO9001系列的两个医疗器械质量管理体系标准,即ISO13485:1996版与ISO13481:1996版,并随9001版本升级不断调整优化,最终在2016年发布了全新的ISO13485:2016版标准。
上海医疗检测设备展|医疗器械注册自检管理规定,那些不知道的细节要点在这里

中国医疗器械行业协会就新修订《医疗器械监督管理条例》及配套规章(上市前)举办了专场培训会,在会上医疗器械注册司的相关领导对《医疗器械注册自检管理规定》进行了相关政策解读。除了正式解读文件,还有尚不清晰的问题不断呈现,针对未理解的部分,医疗器械注册司对细则进行了详细解读。