



我国对医疗器械产品的监管方式主要包括上市前型式检验、注册技术审评、现场体系核查、上市后的监督检查及抽查等。本文梳理分析了近三年以来医疗器械独立软件的技术审评与体系核查中与标准相关的注册发补和体系缺陷项,存在以下几方面的共性问题:
医疗器械独立软件产品在申报过程中,较常见的问题是基本概念不清晰、同一术语在医疗与非医疗领域的具体含义存在差异、产品分类模糊但未提交分类界定告知书、不作为医疗器械管理但提交产品注册申请等问题。人工智能软件产品易出现管理类别(二类或三类)模糊的问题;数字疗法软件、医院信息管理类软件易出现仅将量表数字化、或不具有医疗数据处理功能,因而不作为医疗器械产品管理的问题。若在审评核查过程中发现产品管理类别问题而提交分类界定,或因非医疗器械而撤回注册申请,均会大幅减慢产品的上市速度、耗费企业成本。
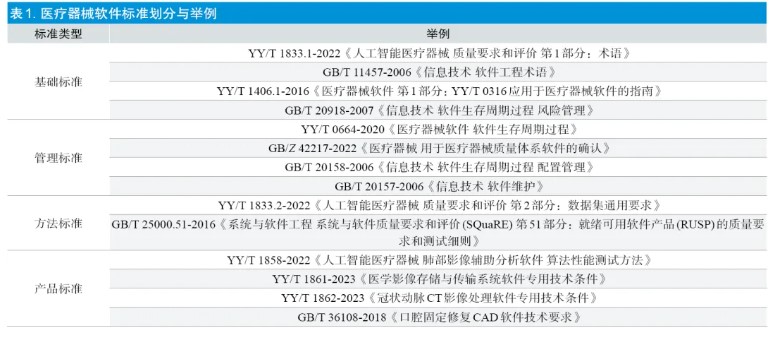
根据《医疗器械软件注册审查指导原则(2022 年修订版)》[1] 的定义,软件产品的性能指标包括通用要求、专用要求、安全要求。其中通用要求根据软件产品特性进行规范,若无专用标准,则由企业自行定义;专用要求是指特定医疗器械软件应符合相关专门的产品标准。技术审评核查中发现,产品技术要求的常见问题包括性能指标不完整、不合理;临床功能描述过于笼统、不准确、不完整;检验方法不合理、不明确,检验方法与标准方法有差异时,未能说明等同性替代的理由;不同企业申报同类型产品的通用要求(尤其是临床功能)的描述差异较大;缺少测试体模或测试工具等必要测试条件信息描述;性能指标和检验方法缺乏研发验证的依据等问题。
通用软件领域的标准并未考虑产品临床使用精度、临床环境下的可靠性等问题[2]。目前,仅治疗计划软件和部分影像处理软件已有产品专用标准,包括YY/T 0895、YY/T0889、YY/T 0798、YY 0721、GB/T 16260.4、YY/T 1858、YY/T 1861 等。除此之外,手术导航及控制系统、生理参数测量设备、心脏节律管理设备等产品,以及数字疗法、虚拟现实、脑机接口等技术均涉及软件,但目前尚无专用的参考标准。部分已发布的软件产品专用标准仅给出软件测试相关的概念性描述,缺乏更为明确的测试规范要求,企业通常需要进一步细化检验方法[3]。此外,部分标准发布时间较长,未及时修订,性能指标及检验方法滞后于现行技术发展。
在现有软件标准框架产品门类覆盖性不足、申报产品缺乏适宜的标准指导的情况下,企业需要结合产品特性自行定义性能指标和检验方法,对于审评核查机构而言,缺乏科学规范的制定依据、检验方法不明确、各企业间差异过大的表述都将导致难以有效对比评价不同软件产品的安全有效性。
当前,我国医疗器械软件质量评价的主要参考标准是GB/T25000.51,检验机构依据该标准运用黑盒测试方法对软件成品开展检测。然而,软件具有非实体性、逻辑严密且缺陷隐蔽性强等特点,易受到测试有限性、退化问题等因素影响,仅通过最终的成品检测无法充分发现软件缺陷,若在实际临床应用中变更频繁,则易引起严重后果,需要对软件的全生命周期进行系统性的评价才能保证产品的安全有效性,这与传统的医疗器械的评价方式存在较大差异[4,5]。
此外,医学数字成像和通信标准(Digital Imaging and Communications in Medicine,DICOM)、卫生信息交换标准(Health Level Seven,HL7)等标准的符合性无须提交测试验证报告,仅通过DICOM符合性声明难以判断标准的实际符合性。以DICOM标准为例,若无验证测试报告,则无法确认申报产品对于医学影像传输的正确性、设备之间通信信息的完整性、患者基本信息及诊断图像显示的一致性、正确性以及有效性等。加之部分软件开发商和用户对数据的标准化存储和保护意识弱,易造成不同产品无法实现互联互通、患者隐私泄露等问题,阻碍了医疗数据的共享与利用。
在算法训练与测试方面,基于深度学习的医疗器械软件对数据集的数量和质量均有较高的要求,但公开数据集通常无法满足开发和性能评价的需求。企业需要通过筛选大量医疗数据来自行构建数据集,耗费大量的时间和人力,此外,数据标注等过程受主客观因素影响较多,较难评价数据集的质量。
临床评价是医疗器械软件的确认环节,若通过同品种医疗器械临床数据进行分析评价的方式开展临床评价,常见问题包括所选择的对比产品与申报产品的算法原理差异较大,对比过于简单或未覆盖必要的功能模块,未提交差异部分的支持性资料等[6]。
软件生存周期过程主要包括软件开发过程、软件维护过程、软件风险管理过程、软件配置管理过程、软件缺陷管理过程。现场核查发现,软件生存周期过程主要如下方面的问题:需求内容不完整,未覆盖独立软件产品风险分析、相关标准要求、已实现的功能模块等内容;设计要求不明确、未根据软件需求实施软件设计;测试与验证方面,软件测试用例过于简单、可操作性差、使用不规范,测试验证未覆盖性能指标和功能模块或风险项、测试记录不完整;未依据企业自定的版本命名规则实施版本迭代控制及配置管理;缺乏缺陷分析修复、回归测试等缺陷管理活动记录。上述问题的可能原因是部分生产企业对软件生命周期过程管理不重视或者理解存在误区,造成标准落实不足、相关活动的记录缺失或不完整。
YY/T 0664/IEC 62304 并未限定软件产品的生存周期模型,企业可根据产品需求、风险特点、开发目标和资源等要素,选择适宜的生存周期模型。相较传统开发模型而言,敏捷开发具有快捷、轻量的优点,需要建立合理的机制来有效地响应变化,重点关注软件更新、文件与记录的有效控制,美国医疗仪器促进协会(The Association for the Advancement of Medical Instrumentation,AAMI)已发布相关指南,但我国目前在医疗器械领域并未建立相关标准,绝大部分生产企业仍遵循瀑布模型和V模型,甚至出现为符合上述模型而编造记录导致设计开发过程的真实性存疑、未能通过现场核查的问题[7]。标准的空缺在一定程度上限制了产品的开发迭代以及行业的发展。
目前我国已发布的涉及生存周期的标准有YY/T 0664、GB/Z 42217、GB/T 20158、GB/T 20157 等,但是在现场核查过程中发现软件生存周期过程管理仍然存在落实不足的问题,建议:①进一步加强软件标准宣贯,统一认识与执行尺度,促进标准的理解与落实;同时建议生产企业严格执行软件全生命周期过程管理;②加强软件变更过程控制,所有变更及验证记录均应充分、完整、可追溯,降低引入新缺陷的风险。同时,建议考虑制定敏捷开发相关过程控制标准。
想要了解更多请前往医疗质量检测技术及测试仪器展
文章来源:中国医疗器械信息
若涉及侵权,请立刻联系删除
关键字:


