



Part.03
无菌包装质量控制
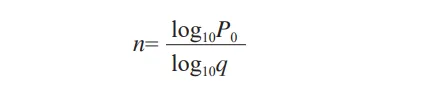
P0——未被发现的风险

确认过程中,除了使用极限参数生产样品进行验证,还要验证不同模具、不同封条对包装的影响。灭菌过程也是影响封口质量的一个因素,灭菌前和灭菌后的封口质量对比也要充分考虑到。还要考虑批量的验证,包括不同人员在不同时间段,生产不同批次的包装产生的差异性。
Part.04
总 结
文章来源:
若涉及侵权,请立刻联系删除
关键字:
Part.03
无菌包装质量控制
P0——未被发现的风险
确认过程中,除了使用极限参数生产样品进行验证,还要验证不同模具、不同封条对包装的影响。灭菌过程也是影响封口质量的一个因素,灭菌前和灭菌后的封口质量对比也要充分考虑到。还要考虑批量的验证,包括不同人员在不同时间段,生产不同批次的包装产生的差异性。
Part.04
总 结
文章来源:
若涉及侵权,请立刻联系删除
关键字:



主办方:
主办方: